
의약품 제조업체: AbbVie Inc. (Updated: 2024-07-12)
처방 정보의 주요 내용
VENCLEXTA® (베네토클락스 정제), 경구 투여
미국 최초 승인: 2016
적응증 및 사용
투여량 및 투여 방법
투여 형태 및 강도
정제: 10mg, 50mg, 100mg (3)
경고 및 주의 사항
- 종양 용해 증후군(TLS): TLS를 예상하고 모든 환자에서 위험을 평가하십시오. 항고요산혈증제로 사전 투여하고 적절한 수분 공급을 유지하십시오. 전반적인 위험이 증가함에 따라 더 강력한 조치(정맥 수액, 빈번한 모니터링, 입원)를 취하십시오. (2.4, 5.1)
- 호중구 감소증: 혈액 수치를 모니터링하십시오. 투여를 중단하고 동일하거나 감소된 용량으로 재개하십시오. 지지 치료 조치를 고려하십시오. (2.5, 5.2)
- 감염: 감염의 징후와 증상을 모니터링하고 즉시 치료하십시오. 3등급 및 4등급 감염의 경우 해결될 때까지 투여를 중단하고 동일하거나 감소된 용량으로 재개하십시오. (2.5, 5.3)
- 면역 접종: B 세포 회복이 될 때까지 VENCLEXTA 치료 전, 중 또는 후에 생백신을 투여하지 마십시오. (5.4)
- 태아 독성: 태아에게 해를 끼칠 수 있습니다. 임신 가능성이 있는 여성에게 태아에 대한 잠재적 위험을 알리고 효과적인 피임을 사용하도록 조언하십시오. (5.5)
- 보르테조밉과 덱사메타손과 병용하여 VENCLEXTA로 다발성 골수종 환자를 치료하는 것은 통제된 임상 시험 외부에서는 권장되지 않습니다. (5.6)
부작용
CLL/SLL에서 오비누투주맙 또는 리툭시맙과 병용 또는 단독 요법으로 VENCLEXTA를 투여했을 때 가장 흔한 부작용(≥20%)은 호중구 감소증, 혈소판 감소증, 빈혈, 설사, 메스꺼움, 상기도 감염, 기침, 근골격계 통증, 피로 및 부종입니다. (6.1)
AML에서 아자시티딘 또는 데시타빈 또는 저용량 시타라빈과 병용했을 때 가장 흔한 부작용(≥30%)은 메스꺼움, 설사, 혈소판 감소증, 변비, 호중구 감소증, 발열성 호중구 감소증, 피로, 구토, 부종, 발열, 폐렴, 호흡 곤란, 출혈, 빈혈, 발진, 복통, 패혈증, 근골격계 통증, 현기증, 기침, 인후통 및 저혈압입니다. (6.1)
의심되는 부작용을 보고하려면 AbbVie Inc.에 1-800-633-9110 또는 FDA에 1-800-FDA-1088 또는 www.fda.gov/medwatch로 연락하십시오.
약물 상호 작용
특정 인구 집단에서의 사용
환자 상담 정보 및 약물 안내는 17을 참조하십시오.
개정: 2024년 7월
목차
전문 정보: 목차*
1 적응증 및 사용법
1.1 만성 림프구성 백혈병/소림프구성 림프종
1.2 급성 골수성 백혈병
2 용법 및 용량
2.1 중요한 안전 정보
2.2 만성 림프구성 백혈병/소림프구성 림프종에 대한 권장 용량
2.3 급성 골수성 백혈병에 대한 권장 용량
2.4 종양 용해 증후군에 대한 위험 평가 및 예방
2.5 이상 반응에 대한 용량 조절
2.6 약물 상호 작용에 대한 용량 조절
2.7 중증 간 기능 장애 환자에 대한 용량 조절
2.8 투여
3 제형 및 강도
4 금기 사항
5 경고 및 주의 사항
5.1 종양 용해 증후군
5.2 호중구 감소증
5.3 감염
5.4 예방 접종
5.5 배아-태아 독성
5.6 VENCLEXTA를 보르테조밉 및 덱사메타손에 추가했을 때 다발성 골수종 환자의 사망률 증가
6 이상 반응
6.1 임상 시험 경험
7 약물 상호 작용
7.1 다른 약물의 VENCLEXTA에 대한 영향
7.2 VENCLEXTA의 다른 약물에 대한 영향
8 특정 인구 집단에서의 사용
8.1 임신
8.2 수유
8.3 생식 능력이 있는 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 신장 장애
8.7 간 기능 장애
10 과량 투여
11 설명
12 약리학
12.1 작용 기전
12.2 약력학
12.3 약동학
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식 능력 저해
13.2 동물 독성 및/또는 약리학
14 임상 연구
14.1 만성 림프구성 백혈병/소림프구성 림프종
14.2 급성 골수성 백혈병
16 포장 단위/보관 및 취급
17 환자 상담 정보
- *
- 전문 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1 적응증 및 용법
1.2 급성 골수성 백혈병
VENCLEXTA는 75세 이상 또는 집중 유도 화학 요법을 사용할 수 없는 동반 질환이 있는 성인의 새로 진단된 급성 골수성 백혈병(AML) 치료에 아자시티딘 또는 데시타빈 또는 저용량 시타라빈과 병용하여 사용됩니다.
2 투여 및 관리
2.1 중요 안전 정보
VENCLEXTA 첫 투여 전에 종양 용해 증후군(TLS) 위험 수준에 대한 환자 특정 요인을 평가하고 TLS 위험을 줄이기 위해 환자에게 예방적 수분 공급 및 항고요산혈증제를 제공하십시오 [용량 및 투여 (2.4) 및 경고 및 주의 사항 (5.1) 참조].
2.2 만성 림프구성 백혈병/소림프구성 림프종에 대한 권장 용량
VENCLEXTA 투여는 5주간의 증량 기간으로 시작됩니다. 5주간의 증량 투여 일정은 종양 부담을 점차적으로 줄이고(감소) TLS 위험을 줄이도록 설계되었습니다.
VENCLEXTA 5주간 용량 증량 일정
표 1에 표시된 바와 같이 5주간의 증량 투여 일정에 따라 VENCLEXTA를 1일 1회 경구로 400mg의 권장 용량으로 투여하십시오.
| VENCLEXTA 경구 일일 용량 |
|
| 1주차 | 20mg |
| 2주차 | 50mg |
| 3주차 | 100mg |
| 4주차 | 200mg |
| 5주차 이후 | 400mg |
CLL/SLL 시작 패키지는 증량 일정에 따라 VENCLEXTA의 첫 4주를 제공합니다 [제공 형태/보관 및 취급 (16) 참조].
오비누투주맙과 병용
1주기 1일 100mg으로 오비누투주맙 투여를 시작한 다음 1주기 2일 900mg을 투여합니다. 1주기 8일과 15일에 1000mg을 투여하고 6주기 동안 총 28일마다 각 주기의 1일에 투여합니다. 추가 투여 정보는 오비누투주맙 처방 정보를 참조하십시오.
1주기 22일에 5주간의 증량 투여 일정에 따라 VENCLEXTA를 시작합니다( 표 1 참조). 2주기 28일에 증량 단계를 완료한 후 12주기의 마지막 날까지 3주기 1일부터 1일 1회 경구로 400mg의 VENCLEXTA를 계속 투여합니다.
리툭시맙과 병용
환자가 VENCLEXTA에 대한 5주간의 증량 투여 일정을 완료하고( 표 1 참조) 1일 1회 경구로 400mg의 VENCLEXTA를 7일 동안 투여한 후 리툭시맙 투여를 시작합니다. 6주기 동안 28일마다 각 주기의 1일에 리툭시맙을 투여하며, 1주기에는 정맥 주사로 375mg/m2, 2-6주기에는 정맥 주사로 500mg/m2의 용량으로 투여합니다. 리툭시맙 1주기 1일부터 24개월 동안 1일 1회 경구로 400mg의 VENCLEXTA를 계속 투여합니다.
추가 투여 정보는 리툭시맙 처방 정보를 참조하십시오.
단독 요법
5주간의 증량 투여 일정을 완료한 후( 표 1 참조) VENCLEXTA의 권장 용량은 1일 1회 400mg입니다. 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 VENCLEXTA를 계속 투여합니다.
2.3 급성 골수성 백혈병에 대한 권장 용량
VENCLEXTA의 권장 용량 및 증량은 병용 약물에 따라 다릅니다. 표 2에 표시된 바와 같이 3일 또는 4일 용량 증량을 포함한 투여 일정을 따르십시오. 병용하여 1주기 1일에 VENCLEXTA 투여를 시작합니다.
- 아자시티딘 75mg/m2를 28일마다 각 주기의 1-7일에 정맥 주사 또는 피하 주사로 1일 1회 투여; 또는
- 데시타빈 20mg/m2를 28일마다 각 주기의 1-5일에 정맥 주사로 1일 1회 투여; 또는
- 시타라빈 20mg/m2를 28일마다 각 주기의 1-10일에 피하 주사로 1일 1회 투여.
| VENCLEXTA 경구 일일 용량 |
||
| 1일 | 100mg | |
| 2일 | 200mg | |
| 3일 | 400mg | |
| 4일 이후 | 28일마다 각 주기의 1일 1회 경구로 400mg 병용하여 아자시티딘 또는 데시타빈 |
28일마다 각 주기의 1일 1회 경구로 600mg 병용하여 저용량 시타라빈 |
2.3 투여 및 관리
아자시티딘 또는 데시타빈 또는 저용량 시타라빈과 병용하여 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 VENCLEXTA를 계속 투여하십시오.
추가 투여 정보는 임상 연구 (14.2) 및 아자시티딘, 데시타빈 또는 시타라빈의 처방 정보를 참조하십시오.
2.4 종양 용해 증후군에 대한 위험 평가 및 예방
VENCLEXTA로 치료받는 환자는 종양 용해 증후군(TLS)이 발생할 수 있습니다. 관리에 대한 자세한 내용은 아래 해당 섹션을 참조하십시오. TLS 위험 수준에 대한 환자 특정 요인을 평가하고 VENCLEXTA 첫 번째 투여 전에 환자에게 예방적 수분 공급 및 항고요산혈증제를 제공하여 TLS 위험을 줄이십시오.
만성 림프구성 백혈병/소림프구성 림프종
VENCLEXTA는 종양을 빠르게 감소시킬 수 있으며, 따라서 초기 5주 증량 단계에서 TLS 위험이 있습니다. TLS와 일치하는 혈액 화학 변화는 VENCLEXTA 첫 번째 투여 후 6~8시간 만에, 그리고 각 투여량 증가 시에 조기에 발생할 수 있습니다. TLS는 투여 중단 후 VENCLEXTA 재개 시에도 발생할 수 있습니다. 투여 중단 후 VENCLEXTA 투여량 조정에 대한 자세한 내용은 표 4 및 표 5를 참조하십시오.
TLS 위험은 여러 요인, 특히 감소된 신장 기능(크레아티닌 청소율 [CLcr] <80 mL/min) 및 종양 부담에 따라 연속적입니다. 비장 비대는 TLS 위험을 증가시킬 수도 있습니다.
모든 환자에서 방사선 평가(예: CT 스캔)를 포함한 종양 부담 평가를 수행하고, VENCLEXTA 치료 시작 전에 혈액 화학(칼륨, 요산, 인, 칼슘 및 크레아티닌)을 평가하여 기존의 이상을 교정하십시오. 종양 부담이 감소함에 따라 위험이 감소할 수 있습니다. [경고 및 주의 사항 (5.1) 및 특정 인구 집단에서의 사용 (8.6)].
아래 표 3은 임상 시험 데이터에서 종양 부담 결정을 기반으로 VENCLEXTA 치료 중 권장되는 TLS 예방 및 모니터링을 설명합니다. 최종 예방 및 모니터링 일정을 결정하기 전에 모든 환자의 동반 질환을 고려하십시오. 증량 단계 중 1주일 이상 지속되는 투여 중단 후 또는 증량 완료 후 2주 이상 지속되는 투여 중단 후 VENCLEXTA를 재개할 때 TLS 위험을 재평가하십시오. 필요에 따라 예방 및 모니터링을 실시하십시오.
| 종양 부담 | 예방 | 혈액 화학 모니터링c,d |
||
| 수분 공급a | 항- 고요산혈증제b |
설정 및 평가 빈도 |
||
| 낮음 | 모든 LN <5 cm AND ALC <25 x109/L |
경구 (1.5~2 L) |
알로푸리놀 | 외래 환자
|
| 중간 | LN 5~<10 cm 또는 ALC ≥25 x109/L |
경구 (1.5~2 L) 및 추가 정맥 주사 고려 |
알로푸리놀 | 외래 환자
|
| 높음 | LN ≥10 cm 또는 ALC ≥25 x109/L AND LN ≥5 cm |
경구 (1.5~2 L) 및 정맥 주사 (내약성에 따라 150~200 mL/hr) |
알로푸리놀; 기준 요산 수치가 높으면 라스부리케이스 고려 | 입원
외래 환자
|
| ALC = absolute lymphocyte count; CLcr = creatinine clearance; LN = lymph node. a경구 수분 섭취가 불가능한 환자에게는 정맥 수액을 투여하십시오. bVENCLEXTA 투여 시작 2~3일 전에 알로푸리놀 또는 잔틴 산화효소 억제제를 투여하십시오. c혈액 화학 검사(칼륨, 요산, 인, 칼슘 및 크레아티닌)를 평가하고 실시간으로 검토하십시오. dTLS 위험이 있는 환자의 경우, 각 후속 증량 용량 투여 시 6~8시간 및 24시간 후에 혈액 화학 검사를 모니터링하십시오. |
||||
급성 골수성 백혈병
- 모든 환자는 VENCLEXTA 투여 시작 전에 백혈구 수가 25 × 109/L 미만이어야 합니다. 치료 전 세포 감소가 필요할 수 있습니다.
- 첫 VENCLEXTA 투여 전에 모든 환자에게 적절한 수분 공급 및 항고요산혈증제를 포함한 예방 조치를 제공하고 증량 단계 동안 계속 제공합니다.
- VENCLEXTA로 치료 시작 전에 혈액 화학 검사(칼륨, 요산, 인, 칼슘 및 크레아티닌)를 평가하고 기존의 이상을 교정합니다.
- 증량 단계 동안 각 새로운 투여 후 투여 전, 6~8시간 및 최종 투여 후 24시간에 TLS에 대한 혈액 화학 검사를 모니터링합니다.
- TLS 위험 요인이 있는 환자(예: 순환 폭발 세포, 골수에서 백혈병 관여가 높음, 치료 전 젖산 탈수소효소[LDH] 수치 상승 또는 신장 기능 저하)의 경우, 증가된 실험실 모니터링 및 VENCLEXTA 시작 용량 감소를 포함한 추가 조치를 고려합니다.
2.5 부작용에 대한 용량 조절
만성 림프구성 백혈병/소림프구성 림프종
부작용에 대한 VENCLEXTA의 권장 용량 조절은 표 4에 나와 있으며 부작용에 대한 VENCLEXTA의 권장 용량 감소는 표 5에 나와 있습니다.
증량 단계 중 1주일 이상 또는 증량 완료 후 2주일 이상 용량 중단이 발생한 환자의 경우 TLS 위험을 재평가하여 감소된 용량으로 재투여가 필요한지 여부를 결정합니다(예: 증량 일정의 모든 수준 또는 일부 수준) [용량 및 투여(2.2, 2.4) 참조].
| 부작용 | 발생 | 용량 조절 |
| 종양 용해 증후군 | ||
| TLS를 시사하는 혈액 화학 변화 또는 증상 [경고 및 주의 사항(5.1) 참조] | 모든 | 다음 날 투여량을 보류합니다. 마지막 투여 후 24~48시간 이내에 해결되면 동일한 용량으로 재개합니다. |
| 48시간 이상 해결하는 데 걸리는 혈액 화학 변화의 경우 감소된 용량으로 재개합니다(표 5 참조). | ||
| 임상 TLSb의 경우 해결 후 감소된 용량으로 재개합니다(표 5 참조). | ||
| 비혈액학적 부작용 | ||
| 3등급 또는 4등급 비혈액학적 독성 [부작용(6.1) 참조] | 첫 번째 발생 | VENCLEXTA를 중단합니다. 1등급 또는 기준선 수준으로 해결되면 동일한 용량으로 VENCLEXTA를 재개합니다. |
| 두 번째 및 그 이후 발생 | VENCLEXTA를 중단합니다. 해결 후 VENCLEXTA로 치료를 재개할 때 표 5의 용량 감소 지침을 따릅니다. 의사의 재량에 따라 더 큰 용량 감소가 발생할 수 있습니다. |
|
| 혈액학적 부작용 | ||
| 감염 또는 발열을 동반한 3등급 호중구 감소증; 또는 4등급 혈액학적 독성(림프구 감소증 제외) [경고 및 주의 사항(5.2) 참조] | 첫 번째 발생 | VENCLEXTA를 중단합니다. 1등급 또는 기준선 수준으로 해결되면 동일한 용량으로 VENCLEXTA를 재개합니다. |
| 두 번째 및 그 이후 발생 | VENCLEXTA를 중단합니다. 해결 후 VENCLEXTA로 치료를 재개할 때 표 5의 용량 감소 지침을 따릅니다. 의사의 재량에 따라 더 큰 용량 감소가 발생할 수 있습니다. |
|
| 2주일 이상 100mg 미만으로 용량 감소가 필요한 환자의 경우 VENCLEXTA를 중단하는 것을 고려합니다. a부작용은 NCI CTCAE 버전 4.0을 사용하여 등급이 매겨졌습니다. b임상 TLS는 급성 신부전, 심장 부정맥 또는 급사 및/또는 발작과 같은 임상적 결과를 동반한 실험실 TLS로 정의되었습니다. [부작용(6.1) 참조]. |
||
| 중단 시 용량, mg | 재개 용량, mga,b |
| 400 | 300 |
| 300 | 200 |
| 200 | 100 |
| 100 | 50 |
| 50 | 20 |
| 20 | 10 |
| a증량 단계 동안, 용량을 증가시키기 전에 1주일 동안 감소된 용량을 유지합니다. b증량 단계 동안 용량 중단이 1주일을 초과하거나 증량 완료 후 2주일을 초과하는 경우, TLS 위험을 재평가하고 감소된 용량으로 재투여가 필요한지 결정합니다 [용량 및 투여 (2.2), 2.4) 참조]. |
|
급성 골수성 백혈병
빈혈이 해소될 때까지 혈액 수치를 자주 모니터링합니다. 빈혈에 대한 용량 조절 및 중단은 관해 상태에 따라 달라집니다. 부작용에 대한 VENCLEXTA 용량 조절은 표 6에 나와 있습니다.
| 부작용 | 발생 | 용량 조절 |
| 혈액학적 부작용 | ||
| 발열 또는 감염 유무와 관계없이 4등급 호중구 감소증; 또는 4등급 혈소판 감소증 [경고 및 주의 사항 (5.2) 참조] | 관해 달성 전 발생a | 대부분의 경우, 아자시티딘, 데시타빈 또는 저용량 시타라빈과 병용하여 VENCLEXTA를 중단하지 않습니다. 관해 달성 전 빈혈이 발생하기 때문입니다. |
| 관해 달성 후 처음 발생하고 7일 이상 지속 | 아자시티딘, 데시타빈 또는 저용량 시타라빈과 병용하여 VENCLEXTA의 다음 사이클을 지연시키고 혈액 수치를 모니터링합니다. 1등급 또는 2등급으로 해소되면 아자시티딘, 데시타빈 또는 저용량 시타라빈과 병용하여 VENCLEXTA를 동일한 용량으로 재개합니다. |
|
| 관해 달성 후 사이클에서 7일 이상 지속되는 후속 발생 | 아자시티딘, 데시타빈 또는 저용량 시타라빈과 병용하여 VENCLEXTA의 다음 사이클을 지연시키고 혈액 수치를 모니터링합니다. 1등급 또는 2등급으로 해소되면 아자시티딘, 데시타빈 또는 저용량 시타라빈과 병용하여 VENCLEXTA를 동일한 용량으로 재개하고, 각 후속 사이클에서 VENCLEXTA 기간을 7일 단축합니다(예: 28일 대신 21일). |
|
| 비혈액학적 부작용 | ||
| 3등급 또는 4등급 비혈액학적 독성 [부작용 (6.1) 참조] | 모든 발생 | 지지 요법으로 해결되지 않으면 VENCLEXTA를 중단합니다. 1등급 또는 기준선 수준으로 해소되면 VENCLEXTA를 동일한 용량으로 재개합니다. |
| a골수 평가를 권장합니다. | ||
2.6 약물 상호 작용에 대한 용량 조절
강력하거나 중등도의 CYP3A 억제제 또는 P-gp 억제제
표 7은 강력하거나 중등도의 CYP3A 억제제 또는 P-gp 억제제와 동시에 사용하는 경우 VENCLEXTA 금기 사항 또는 용량 조절을 설명합니다 [약물 상호 작용 (7.1)]를 참조하십시오. ramp-up 단계 시작, 진행 또는 종료 시.
강력하거나 중등도의 CYP3A 억제제 또는 P-gp 억제제를 중단한 후 2~3일 후에 동시 사용 전에 사용했던 VENCLEXTA 용량을 재개하십시오 [약물 상호 작용 (7.1)]를 참조하십시오.
| 동시 투여 약물 |
시작 및 ramp-up 단계 |
일일 안정 용량 (ramp-up 단계 후)a |
|
| 포사코나졸 | CLL/SLL | 금기 | VENCLEXTA 용량을 70mg으로 줄입니다. |
| AML | 1일차 – 10mg 2일차 – 20mg 3일차 – 50mg 4일차 – 70mg |
||
| 기타 강력한 CYP3A 억제제 |
CLL/SLL | 금기 | VENCLEXTA 용량을 100mg으로 줄입니다. |
| AML | 1일차 – 10mg 2일차 – 20mg 3일차 – 50mg 4일차 – 100mg |
||
| 중등도 CYP3A 억제제 |
VENCLEXTA 용량을 최소 50% 줄입니다. | ||
| P-gp 억제제 | |||
| aCLL/SLL 환자의 경우 표 7에 설명된 대로 대체 약물을 고려하거나 VENCLEXTA 용량을 줄입니다. | |||
2.7 중증 간 기능 장애 환자에 대한 용량 조절
중증 간 기능 장애(Child-Pugh C) 환자의 경우 VENCLEXTA 1일 1회 용량을 50% 줄입니다. 이러한 환자의 경우 이상 반응을 더 자주 모니터링하십시오 [특정 인구 집단에서의 사용 (8.7)]를 참조하십시오.
2.8 투여
환자에게 다음과 같이 지시하십시오.
- VENCLEXTA를 식사와 함께 물과 함께 복용하십시오.
- VENCLEXTA를 매일 거의 같은 시간에 복용하십시오.
- VENCLEXTA 정제를 통째로 삼키십시오. 삼키기 전에 정제를 씹거나, 부수거나, 쪼개지 마십시오.
VENCLEXTA의 권장 용량은 승인된 정제 강도를 사용하여 투여할 수 있습니다(예: 필요에 따라 1 x 100mg 정제 대신 2 x 50mg 정제 또는 10 x 10mg 정제를 복용할 수 있음).
환자가 VENCLEXTA 용량을 복용한 후 8시간 이내에 용량을 놓친 경우 환자에게 가능한 한 빨리 놓친 용량을 복용하고 정상적인 일일 복용 일정을 재개하도록 지시하십시오. 환자가 8시간 이상 용량을 놓친 경우 환자에게 놓친 용량을 복용하지 말고 다음 날 평소 복용 일정을 재개하도록 지시하십시오.
환자가 복용 후 구토를 하는 경우 환자에게 그날 추가 용량을 복용하지 말고 다음에 처방된 용량을 평소 시간에 복용하도록 지시하십시오.
3 제형 및 함량
| Tablet Strength | Description of Tablet |
| 10 mg | 한쪽 면에는 “V”, 다른 쪽 면에는 “10”이 각인된 둥글고 양쪽 볼록한 연한 노란색 필름 코팅 정제 |
| 50 mg | 한쪽 면에는 “V”, 다른 쪽 면에는 “50”이 각인된 타원형, 양쪽 볼록한 베이지색 필름 코팅 정제 |
| 100 mg | 한쪽 면에는 “V”, 다른 쪽 면에는 “100”이 각인된 타원형, 양쪽 볼록한 연한 노란색 필름 코팅 정제 |
4 금기사항
CLL/SLL 환자에서 VENCLEXTA와 강력한 CYP3A 억제제를 초기 및 증량 단계에서 병용하는 것은 종양 용해 증후군 위험 증가 가능성으로 인해 금기입니다 [용법 및 용량 (2.6) 및 약물 상호 작용 (7.1) 참조].
5 경고 및 주의사항
5.1 종양 용해 증후군
투석이 필요한 치명적인 사건 및 신부전을 포함한 종양 용해 증후군(TLS)이 VENCLEXTA로 치료받은 환자에게 발생했습니다 [유해 반응 (6.1)] 참조.
VENCLEXTA는 종양의 빠른 감소를 유발할 수 있으며, 따라서 모든 환자의 투여 시작 및 증량 단계에서, 그리고 CLL/SLL 환자의 투여 중단 후 재투여 시 TLS 위험이 있습니다. TLS와 일치하는 혈액 화학 변화는 VENCLEXTA 첫 번째 투여 후 6~8시간 만에, 그리고 각 용량 증가 시에 발생할 수 있습니다. VENCLEXTA 20mg 단일 투여 후 치명적인 경우를 포함한 TLS가 보고되었습니다.
현재(5주) 용량 증량 및 TLS 예방 및 모니터링 조치를 따른 CLL/SLL 환자의 경우 VENCLEXTA CLL/SLL 단독 요법 시험에서 TLS 발생률은 2%였습니다. TLS 발생률은 오비누투주맙 또는 리툭시맙과 병용한 VENCLEXTA에서 일관되었습니다. CLL/SLL 환자의 경우 2~3주 용량 증량 및 더 높은 시작 용량으로 TLS 발생률은 13%였으며, 사망 및 신부전이 포함되었습니다 [유해 반응 (6.1)] 참조.
현재 3일 증량 투약 일정 및 TLS 예방 및 모니터링 조치를 따른 AML 환자의 경우 아자시티딘과 병용한 VENCLEXTA를 투여받은 환자에서 TLS 발생률은 1.1%였습니다(VIALE-A). 4일 증량 투약 일정 및 TLS 예방 및 모니터링 조치를 따른 AML 환자의 경우 저용량 시타라빈과 병용한 VENCLEXTA를 투여받은 환자에서 TLS 발생률은 5.6%였으며, 사망 및 신부전이 포함되었습니다 [유해 반응 (6.1)] 참조.
TLS 위험은 여러 요인, 특히 감소된 신장 기능, 종양 부담 및 악성 종양 유형에 따라 연속적입니다. CLL/SLL 환자의 경우 비장 비대가 TLS 위험을 증가시킬 수도 있습니다.
모든 환자의 위험을 평가하고 수분 공급 및 항고요산혈증제를 포함한 TLS에 대한 적절한 예방 조치를 취하십시오. 혈액 화학을 모니터링하고 이상을 신속하게 관리하십시오. 전반적인 위험이 증가함에 따라 더 강력한 조치(정맥 수액, 빈번한 모니터링, 입원)를 사용하십시오. 필요한 경우 투약을 중단하십시오. VENCLEXTA를 재투여할 때는 용량 조정 지침을 따르십시오 [용법 및 투약 (2.1, 2.2, 2.3, 2.4) 및 특정 인구 집단에서의 사용 (8.6)] 참조.
P-gp 억제제 또는 강력하거나 중등도의 CYP3A 억제제와 VENCLEXTA를 병용하면 베네토클락 노출이 증가하여 VENCLEXTA 투여 시작 및 증량 단계에서 TLS 위험이 증가할 수 있습니다. CLL/SLL 환자의 경우 투여 시작 및 5주 증량 단계에서 강력한 CYP3A 억제제와 VENCLEXTA를 병용하는 것은 금기입니다 [금기 사항 (4)] 참조. AML 환자의 경우 투여 시작 및 3일 또는 4일 증량 단계에서 강력한 CYP3A 억제제와 병용할 때 VENCLEXTA 용량을 줄이십시오. CLL/SLL 또는 AML 환자의 경우 중등도 CYP3A4 억제제 또는 P-gp 억제제와 병용할 때 VENCLEXTA 용량을 줄이십시오 [용법 및 투약 (2.6) 및 약물 상호 작용 (7.1)] 참조.
5.2 호중구 감소증
CLL 환자의 경우 VENCLEXTA 병용 및 단독 요법 연구에서 VENCLEXTA로 치료받은 환자의 63~64%에서 3등급 또는 4등급 호중구 감소증이 발생했고, 31~33%에서 4등급 호중구 감소증이 발생했습니다. 발열성 호중구 감소증은 4~6%의 환자에게 발생했습니다 [유해 반응 (6.1)] 참조.
AML 환자의 경우 아자시티딘, 데시타빈 또는 저용량 시타라빈과 병용한 VENCLEXTA로 치료받은 환자의 95~100%에서 기준 호중구 수가 악화되었습니다. 호중구 감소증은 후속 사이클에서 재발할 수 있습니다.
치료 기간 동안 전체 혈구 수를 모니터링하십시오. 심각한 호중구 감소증에 대한 VENCLEXTA 중단 및 재투여에 대해서는 CLL의 경우 표 4, AML의 경우 표 6을 참조하십시오 [용법 및 투약 (2.5)] 참조. 항생제 및 성장 인자(예: G-CSF)를 포함한 지지 치료를 고려하십시오.
5.3 감염
폐렴 및 패혈증과 같은 치명적인 감염 및 심각한 감염이 VENCLEXTA로 치료받은 환자에게 발생했습니다 [유해 반응 (6.1)] 참조.
환자의 감염 징후 및 증상을 모니터링하고 신속하게 치료하십시오. 3등급 및 4등급 감염의 경우 감염이 해결될 때까지 VENCLEXTA를 중단하십시오. 용량 재투여에 대해서는 CLL의 경우 표 4, AML의 경우 표 6을 참조하십시오 [용법 및 투약 (2.5)] 참조.
5.4 면역
B 세포 회복이 일어날 때까지 VENCLEXTA 치료 전, 중 또는 후에 생백신을 투여하지 마십시오. VENCLEXTA 치료 중 또는 후에 생백신으로 면역을 시행한 경우 안전성 및 유효성은 연구되지 않았습니다. 환자에게 백신 접종의 효과가 떨어질 수 있다고 알려주십시오.
5.5 배아-태아 독성
동물 연구 결과 및 작용 기전을 고려할 때, VENCLEXTA는 임산부에게 투여하면 배아-태아에 해를 끼칠 수 있습니다. 임신한 동물에게 매일 400mg 용량으로 투여한 환자에서 관찰된 것과 동일한 노출량으로 베네토클락스를 투여한 배아-태아 연구에서 착상 후 손실 및 태아 체중 감소가 발생했습니다.
임산부에게 태아에 대한 잠재적 위험을 알려주십시오. 임신 가능성이 있는 여성에게는 VENCLEXTA 치료 중 및 마지막 투여 후 30일 동안 효과적인 피임 방법을 사용하도록 조언하십시오 [특정 집단에서의 사용 (8.1, 8.3)]을 참조하십시오.
5.6 VENCLEXTA를 보르테조밉 및 덱사메타손에 추가했을 때 다발성 골수종 환자의 사망률 증가
재발성 또는 불응성 다발성 골수종 환자를 대상으로 한 무작위 대조 시험(BELLINI; NCT02755597)에서 VENCLEXTA를 보르테조밉 및 덱사메타손에 추가한 경우, VENCLEXTA가 적응증이 아닌 용도로 사용되었으며 사망률이 증가했습니다. VENCLEXTA를 보르테조밉 및 덱사메타손과 병용하여 다발성 골수종 환자를 치료하는 것은 통제된 임상 시험 외부에서는 권장되지 않습니다.
6 부작용
다음 임상적으로 중요한 유해 반응은 라벨링의 다른 부분에서 설명되어 있습니다.
6.1 임상 시험 경험
임상 시험은 매우 다양한 조건에서 수행되므로, 약물의 임상 시험에서 관찰된 유해 사건 발생률은 다른 약물의 임상 시험에서 관찰된 발생률과 직접 비교할 수 없으며, 실제로 관찰된 발생률을 반영하지 않을 수 있습니다.
CLL/SLL에서 안전성 모집단은 M13-982, M14-032 및 M12-175에서 환자의 단독 요법으로 VENCLEXTA에 대한 노출과 CLL14 및 MURANO에서 환자의 오비누투주맙 또는 리툭시맙과의 병용 요법을 반영합니다. 이 CLL/SLL 안전성 모집단에서 VENCLEXTA에 대한 가장 흔한 유해 반응(≥20%)은 호중구 감소증, 혈소판 감소증, 빈혈, 설사, 메스꺼움, 상기도 감염, 기침, 근골격계 통증, 피로 및 부종이었습니다.
AML에서 안전성 모집단은 M14-358, VIALE-A 및 VIALE-C에서 환자의 데시타빈, 아자시티딘 또는 저용량 시타라빈과의 병용 요법으로 VENCLEXTA에 대한 노출을 반영합니다. 이 안전성 모집단에서 가장 흔한 유해 반응(임상 시험에서 ≥30%)은 메스꺼움, 설사, 혈소판 감소증, 변비, 호중구 감소증, 발열성 호중구 감소증, 피로, 구토, 부종, 발열, 폐렴, 호흡 곤란, 출혈, 빈혈, 발진, 복통, 패혈증, 근골격계 통증, 현기증, 기침, 인후통 및 저혈압이었습니다.
만성 림프구성 백혈병/소림프구성 림프종
오비누투주맙과 병용한 VENCLEXTA
오비누투주맙과 병용한 VENCLEXTA(VEN+G)(N=212)의 안전성은 이전에 치료를 받지 않은 CLL 환자를 대상으로 한 무작위 배정, 공개, 적극적인 대조군 임상 시험인 CLL14에서 오비누투주맙과 클로람부실의 병용 요법(GClb)(N=214)과 비교하여 평가되었습니다. [임상 연구 (14.1)]. VEN+G군에 무작위 배정된 환자는 6주기 동안 VENCLEXTA와 오비누투주맙을 병용 요법으로 치료받았고, 그 후 6주기 동안 VENCLEXTA를 단독 요법으로 치료받았습니다. 환자는 1주기 22일에 VENCLEXTA의 5주 증량을 시작했고, 완료되면 총 12주기 동안 매일 1회 경구로 VENCLEXTA 400mg을 계속 복용했습니다. 이 임상 시험은 총 누적 질병 평가 척도(CIRS) >6 또는 CLcr <70 mL/min, 간 트랜스아미나제 및 총 빌리루빈 ≤ 정상 상한의 2배를 요구했으며, 눈, 귀, 코 및 인후 기관계를 제외한 CIRS에 의한 개별 기관/시스템 장애 점수가 4인 환자는 제외했습니다. VENCLEXTA에 대한 중간 노출 기간은 10.5개월(범위: 0~13.5개월)이었고, 오비누투주맙의 중간 주기 수는 VEN+G군에서 6이었습니다.
중증 유해 반응은 VEN+G군 환자의 49%에서 보고되었으며, 가장 흔한 원인은 발열성 호중구 감소증과 폐렴(각각 5%)이었습니다. 질병 진행 없이 발생하고 마지막 연구 치료 후 28일 이내에 발생한 치명적인 유해 반응은 환자의 2%(4/212)에서 보고되었으며, 가장 흔한 원인은 감염이었습니다.
VEN+G군에서 유해 반응으로 인해 16%의 환자에서 치료 중단, 21%의 환자에서 용량 감소, 74%의 환자에서 용량 중단이 발생했습니다. 호중구 감소증으로 인해 2%의 환자에서 VENCLEXTA 중단, 13%의 환자에서 용량 감소, 41%의 환자에서 용량 중단이 발생했습니다.
표 9는 CLL14에서 확인된 유해 반응을 보여줍니다.
| 유해 반응 | VENCLEXTA + 오비누투주맙 (N = 212) |
오비누투주맙 + 클로람부실 (N = 214) |
||
| 모든 등급 (%) |
등급 ≥3 (%) |
모든 등급 (%) |
등급 ≥3 (%) |
|
| 혈액 및 림프계 장애 | ||||
| 호중구 감소증a | 60 | 56 | 62 | 52 |
| 빈혈a | 17 | 8 | 20 | 7 |
| 위장관 장애 | ||||
| 설사 | 28 | 4 | 15 | 1 |
| 메스꺼움 | 19 | 0 | 22 | 1 |
| 변비 | 13 | 0 | 9 | 0 |
| 구토 | 10 | 1 | 8 | 1 |
| 일반적인 장애 및 투여 부위 상태 | ||||
| 피로a | 21 | 2 | 23 | 1 |
| 감염 및 기생충 감염 | ||||
| 상기도 감염 a |
17 | 1 | 17 | 1 |
| a여러 가지 이상 반응 용어를 포함합니다. | ||||
VEN+G로 치료받은 환자의 10% 미만에서 보고된 다른 임상적으로 중요한 이상 반응(모든 등급)은 다음과 같습니다.
혈액 및 림프계 장애: 발열성 호중구 감소증(6%)
감염 및 기생충 감염 (모두 여러 가지 이상 반응 용어를 포함합니다.): 폐렴(9%), 요로 감염(6%), 패혈증(4%)
대사 및 영양 장애: 종양 용해 증후군(1%)
VEN+G 완료 후 VENCLEXTA 단독 요법으로 치료하는 동안 환자의 10% 이상에서 발생한 이상 반응은 호중구 감소증(26%)이었습니다. 환자의 2% 이상에서 발생한 3등급 이상의 이상 반응은 호중구 감소증(23%)과 빈혈(2%)이었습니다.
표 10은 CLL14에서 실험실 이상을 보여줍니다.
| 실험실 이상a | VENCLEXTA + 오비누투주맙 (N = 212) |
오비누투주맙 + 클로람부실 (N = 214) |
||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 혈액학 | ||||
| 백혈구 감소증 | 90 | 46 | 89 | 41 |
| 림프구 감소증 | 87 | 57 | 87 | 51 |
| 호중구 감소증 | 83 | 63 | 79 | 56 |
| 혈소판 감소증 | 68 | 28 | 71 | 26 |
| 빈혈 | 53 | 15 | 46 | 11 |
| 화학 | ||||
| 혈액 크레아티닌 증가 | 80 | 6 | 74 | 2 |
| 저칼슘혈증 | 67 | 9 | 58 | 4 |
| 고칼륨혈증 | 41 | 4 | 35 | 3 |
| 고요산혈증 | 38 | 38 | 38 | 38 |
| a새롭게 발생하거나 악화된 실험실 이상 또는 기준선에서 악화된 정도를 알 수 없는 실험실 이상을 포함합니다. | ||||
VEN+G로 치료받은 환자의 2% 이상에서 발생한 4등급 실험실 이상은 호중구 감소증(32%), 백혈구 감소증 및 림프구 감소증(10%), 혈소판 감소증(8%), 저칼슘혈증(8%), 고요산혈증(7%), 혈청 크레아티닌 증가(3%), 고칼슘혈증(3%), 저칼륨혈증(2%)이었습니다.
리툭시맙과 병용 투여한 VENCLEXTA
리툭시맙과 병용 투여한 VENCLEXTA(VEN+R)(N=194)의 안전성은 MURANO에서 벤다무스틴과 리툭시맙 병용 투여(B+R)(N=188)와 비교 평가되었습니다. [임상 연구(14.1)] VEN+R에 무작위 배정된 환자는 예정된 증량(5주)을 완료하고 리툭시맙과 병용하여 VENCLEXTA 400mg을 1일 1회 투여받았으며, 증량 후 총 24개월 동안 VENCLEXTA 단독 요법을 받았습니다. 분석 시점에서 VENCLEXTA에 대한 중간 노출 기간은 22개월이었고, VEN+R군에서 리툭시맙의 중간 사이클 수는 6개였습니다.
VEN+R군 환자의 46%에서 심각한 이상 반응이 보고되었으며, 가장 흔한(≥5%) 이상 반응은 폐렴(9%)이었습니다. 질병 진행 없이 마지막 VENCLEXTA 치료 후 30일 이내 또는 마지막 리툭시맙 치료 후 90일 이내에 발생한 치명적인 이상 반응은 환자의 2%(4/194)에서 보고되었습니다.
VEN+R군에서 이상 반응으로 인해 16%의 환자가 치료를 중단했고, 15%의 환자가 용량을 감소시켰으며, 71%의 환자가 용량을 중단했습니다. 호중구 감소증과 혈소판 감소증은 각각 환자의 3%에서 VENCLEXTA 중단으로 이어졌습니다. 호중구 감소증은 환자의 46%에서 VENCLEXTA 용량 중단으로 이어졌습니다.
표 11은 MURANO에서 확인된 이상 반응을 보여줍니다.
| 이상 반응 | VENCLEXTA + 리툭시맙 (N = 194) |
벤다무스틴 + 리툭시맙 (N = 188) |
||
| 모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
| 혈액 및 림프계 장애 | ||||
| 호중구 감소증a | 65 | 62 | 50 | 44 |
| 빈혈a | 16 | 11 | 23 | 14 |
| 위장관 장애 | ||||
| 설사 | 40 | 3 | 17 | 1 |
| 오심 | 21 | 1 | 34 | 1 |
| 변비 | 14 | <1 | 21 | 0 |
| 감염 및 기생충 감염 | ||||
| 상기도 감염a | 39 | 2 | 23 | 2 |
| 하기도 감염a | 18 | 2 | 10 | 2 |
| 폐렴a | 10 | 7 | 14 | 10 |
| 일반적인 장애 및 투여 부위 상태 | ||||
| 피로a | 22 | 2 | 26 | <1 |
| a여러 이상 반응 용어를 포함합니다. | ||||
VEN+R로 치료받은 환자의 10% 미만에서 보고된 다른 임상적으로 중요한 유해 반응(모든 등급)은 다음과 같습니다.
혈액 및 림프계 장애: 발열성 호중구 감소증(4%)
위장 장애: 구토(8%)
감염 및 기생충 감염: 패혈증(<1%)
대사 및 영양 장애: 종양 용해 증후군(3%)
VEN+R 병용 치료 완료 후 VENCLEXTA 단독 요법으로 치료하는 동안 환자의 ≥10%에서 발생한 유해 반응은 상기도 감염(21%), 설사(19%), 호중구 감소증(16%) 및 하기도 감염(11%)이었습니다. 환자의 ≥2%에서 발생한 3등급 또는 4등급 유해 반응은 호중구 감소증(12%) 및 빈혈(3%)이었습니다.
표 12는 MURANO에서 확인된 실험실 이상을 보여줍니다.
| 실험실 이상 | VENCLEXTA + 리툭시맙 (N = 194) |
벤다무스틴 + 리툭시맙 (N = 188) |
||
| 모든 등급a (%) |
3등급 또는 4등급 (%) |
모든 등급a (%) |
3등급 또는 4등급 (%) |
|
| 혈액학 | ||||
| 백혈구 감소증 | 89 | 46 | 81 | 35 |
| 림프구 감소증 | 87 | 56 | 79 | 55 |
| 호중구 감소증 | 86 | 64 | 84 | 59 |
| 빈혈 | 50 | 12 | 63 | 15 |
| 혈소판 감소증 | 49 | 15 | 60 | 20 |
| 화학 | ||||
| 혈액 크레아티닌 증가 | 77 | <1 | 78 | 1 |
| 저칼슘혈증 | 62 | 5 | 51 | 2 |
| 고요산혈증 | 36 | 36 | 33 | 33 |
| 고칼륨혈증 | 24 | 3 | 19 | 2 |
| a새로운 또는 악화된 실험실 이상 또는 기준선에서 악화된 정도를 알 수 없는 실험실 이상을 포함합니다. | ||||
VEN+R로 치료받은 환자의 2% 이상에서 발생한 4등급 실험실 이상은 호중구 감소증(31%), 림프구 감소증(16%), 백혈구 감소증(6%), 혈소판 감소증(6%), 고요산혈증(4%), 저칼슘혈증(2%), 저혈당(2%), 고마그네슘혈증(2%)이었습니다.
단독 요법으로 사용되는 VENCLEXTA
VENCLEXTA의 안전성은 3개의 단일군 시험(M13-982, M14-032 및 M12-175)의 통합 데이터에서 평가되었습니다. 환자는 증량 단계를 완료한 후 1일 1회 경구로 VENCLEXTA 400mg을 투여받았습니다(N=352). 데이터 분석 시점에서 VENCLEXTA 치료의 중앙값 기간은 14.5개월(범위: 0~50개월)이었습니다. 환자의 52%가 60주 이상 VENCLEXTA를 투여받았습니다.
통합 데이터 세트에서 중앙값 연령은 66세(범위: 28~85세)였으며, 93%가 백인이었고, 68%가 남성이었습니다. 이전 치료의 중앙값 수는 3(범위: 0~15)이었습니다.
환자의 52%에서 심각한 이상 반응이 보고되었으며, 가장 빈번한(≥5%) 이상 반응은 폐렴(9%), 발열성 호중구 감소증(5%) 및 패혈증(5%)이었습니다. VENCLEXTA 단독 요법 연구에서 질병 진행 없이 VENCLEXTA 치료 후 30일 이내에 발생한 치명적인 이상 반응은 환자의 2%에서 보고되었으며, 가장 흔한(2명의 환자) 원인은 패혈성 쇼크였습니다.
이상 반응으로 인해 환자의 9%에서 치료 중단, 13%에서 용량 감소, 36%에서 용량 중단이 발생했습니다. 약물 중단으로 이어진 가장 빈번한 이상 반응은 혈소판 감소증과 자가 면역 용혈성 빈혈이었습니다. 용량 감소 또는 중단으로 이어진 가장 빈번한 이상 반응(≥5%)은 호중구 감소증(8%)이었습니다.
표 13은 이러한 시험에서 확인된 이상 반응을 보여줍니다.
| 이상 반응 | VENCLEXTA (N = 352) |
|
| 모든 등급 (%) |
3등급 이상 (%) |
|
| 혈액 및 림프계 장애 | ||
| 호중구 감소증a | 50 | 45 |
| 빈혈a | 33 | 18 |
| 혈소판 감소증a | 29 | 20 |
| 림프구 감소증a | 11 | 7 |
| 발열성 호중구 감소증 | 6 | 6 |
| 위장 장애 | ||
| 설사 | 43 | 3 |
| 오심 | 42 | 1 |
| 복통a | 18 | 3 |
| 구토 | 16 | 1 |
| 변비 | 16 | <1 |
| 점막염a | 13 | <1 |
| 감염 및 기생충 감염 | ||
| 상기도 감염a | 36 | 1 |
| 폐렴a | 14 | 8 |
| 하기도 감염a | 11 | 2 |
| 일반적인 장애 및 투여 부위 상태 | ||
| 피로a | 32 | 4 |
| 부종a | 22 | 2 |
| 발열 | 18 | <1 |
| 근골격계 및 결합 조직 장애 | ||
| 근골격계 통증a | 29 | 2 |
| 관절통 | 12 | <1 |
| 호흡기, 흉곽 및 종격 장애 | ||
| 기침a | 22 | 0 |
| 호흡곤란a | 13 | 1 |
| 신경계 장애 | ||
| 두통 | 18 | <1 |
| 현기증a | 14 | 0 |
| 피부 및 피하 조직 장애 | ||
| 발진a | 18 | <1 |
| NCI Common Terminology Criteria for Adverse Events version 4.0을 사용하여 등급이 매겨진 이상 반응. a여러 이상 반응 용어를 포함합니다. |
||
표 14 는 치료 전반에 걸쳐 보고된 기준선에서 새롭거나 악화된 실험실 이상을 보여줍니다. VENCLEXTA 단독 요법에서 관찰된 가장 흔한(>5%) 등급 4 실험실 이상은 호중구 감소증(33%), 백혈구 감소증(11%), 혈소판 감소증(15%) 및 림프구 감소증(9%)을 포함한 혈액학적 실험실 이상이었습니다.
| 실험실 이상 | VENCLEXTA (N = 352) |
|
| 모든 등급a (%) |
등급 3 또는 4 (%) |
|
| 혈액학 | ||
| 백혈구 감소증 | 89 | 42 |
| 호중구 감소증 | 87 | 63 |
| 림프구 감소증 | 74 | 40 |
| 빈혈 | 71 | 26 |
| 혈소판 감소증 | 64 | 31 |
| 화학 | ||
| 저칼슘혈증 | 87 | 12 |
| 고혈당증 | 67 | 7 |
| 고칼륨혈증 | 59 | 5 |
| AST 증가 | 53 | 3 |
| 저알부민혈증 | 49 | 2 |
| 저인산혈증 | 45 | 11 |
| 저나트륨혈증 | 40 | 9 |
| a새롭거나 악화되었거나 기준선에서 악화된 실험실 이상을 포함합니다. | ||
CLL/SLL에서 중요한 유해 반응
종양 용해 증후군
종양 용해 증후군은 VENCLEXTA 투여를 시작할 때 확인된 중요한 위험입니다.
CLL14
VEN+G로 치료받은 환자에서 TLS 발생률은 1%(3/212)였습니다 [경고 및 주의 사항 (5.1)]. TLS 3건 모두 해결되었으며 시험에서 탈락으로 이어지지 않았습니다. TLS 사건에 대한 반응으로 2건에서 오비누투주맙 투여가 지연되었습니다.
MURANO
VEN+R로 치료받은 환자에서 TLS 발생률은 3%(6/194)였습니다. 시험에 77/389명의 환자가 등록된 후, 프로토콜이 수정되어 2.2절과 2.4절에 설명된 현재 TLS 예방 및 모니터링 조치가 통합되었습니다 [투여 및 관리 (2.2, 2.4)]. TLS 사건은 모두 VENCLEXTA 증량 기간 동안 발생했으며 2일 이내에 해결되었습니다. 6명의 환자 모두 증량을 완료하고 VENCLEXTA 권장 일일 용량인 400mg에 도달했습니다. 현재 5주 증량 일정과 TLS 예방 및 모니터링 조치를 따른 환자에서 임상적 TLS는 관찰되지 않았습니다 [투여 및 관리 (2.2, 2.4)]. VEN+R로 치료받은 환자의 TLS와 관련된 실험실 이상의 비율은 표 12에 제시되어 있습니다.
단독 요법 연구 (M13-982 및 M14-032)
2.1절과 2.2절에 설명된 권장 사항에 따라 치료받은 CLL 환자 168명에서 TLS 발생률은 2%였습니다 [투여 및 관리 (2.2, 2.4)]. 모든 사건은 실험실 TLS 기준(서로 24시간 이내에 다음 중 ≥2개의 실험실 이상: 칼륨 >6 mmol/L, 요산 >476 µmol/L, 칼슘 <1.75 mmol/L 또는 인 >1.5 mmol/L)을 충족하거나 TLS 사건으로 보고되었습니다. 이러한 사건은 림프절(들) ≥5cm 및/또는 절대 림프구 수(ALC) ≥25 x 109/L인 환자에게 발생했습니다. 모든 사건은 5일 이내에 해결되었습니다. 이러한 환자에서 급성 신부전, 심장 부정맥 또는 급사 및/또는 발작과 같은 임상적 결과를 동반한 TLS는 관찰되지 않았습니다. 모든 환자의 CLcr은 ≥50 mL/min이었습니다. TLS와 관련된 실험실 이상은 고칼륨혈증(모든 등급의 17%, 등급 ≥3의 1%), 고인산혈증(모든 등급의 14%, 등급 ≥3의 2%), 저칼슘혈증(모든 등급의 16%, 등급 ≥3의 2%) 및 고요산혈증(모든 등급의 10%, 등급 ≥3의 <1%)이었습니다.
증량 단계가 더 짧고(2-3주) 시작 용량이 더 높았던 초기 1상 용량 결정 시험에서 TLS 발생률은 13%(10/77; 실험실 TLS 5건, 임상적 TLS 5건)였으며, 여기에는 치명적인 사건 2건과 급성 신부전 3건(투석이 필요한 1건)이 포함되었습니다. 이러한 경험 이후, TLS 위험 평가, 투약 요법, TLS 예방 및 모니터링 조치가 개정되었습니다 [투여 및 관리 (2.2, 2.4)].
급성 골수성 백혈병
아자시티딘과 병용한 VENCLEXTA
새로 진단된 AML 환자를 대상으로 한 이중맹검, 무작위 배정 시험인 VIALE-A에서 아자시티딘(VEN+AZA)과 병용한 VENCLEXTA(N=283)의 안전성을 위약과 병용한 아자시티딘(PBO+AZA)(N=144)과 비교하여 평가했습니다 [임상 연구 (14.2)]. 기준선에서 환자는 ≥75세였거나 다음 기준 중 하나 이상에 따라 집중적 유도 화학 요법을 사용할 수 없는 동반 질환이 있었습니다. 기준선 ECOG 수행 상태 2-3, 심각한 심장 또는 폐 동반 질환, 중등도 간 기능 장애, CLcr <45 mL/min 또는 기타 동반 질환. 환자는 증량 단계 완료 후 아자시티딘(28일 주기의 1-7일에 정맥 또는 피하로 75 mg/m2)과 병용한 VENCLEXTA 400mg을 경구로 하루에 한 번 또는 위약과 병용한 아자시티딘을 무작위로 배정받았습니다. VEN+AZA를 투여받은 환자 중 VENCLEXTA에 대한 중간 노출 기간은 7.6개월(범위: <0.1~30.7개월)이었습니다.
VEN+AZA를 투여받은 환자의 83%에서 중대한 유해 반응이 보고되었으며, 가장 흔한 반응(≥5%)은 발열성 호중구 감소증(30%), 폐렴(22%), 패혈증(진균 제외; 19%) 및 출혈(6%)이었습니다. VEN+AZA를 투여받은 환자의 23%에서 치명적인 유해 반응이 발생했으며, 가장 흔한 반응(≥2%)은 폐렴(4%), 패혈증(진균 제외; 3%) 및 출혈(2%)이었습니다.
유해 반응으로 인해 환자의 24%에서 VENCLEXTA를 영구적으로 중단했고, 2%에서 용량을 감소시켰으며, 72%에서 용량을 중단했습니다. 환자의 ≥2%에서 VENCLEXTA 중단으로 이어진 유해 반응은 패혈증(진균 제외; 3%)과 폐렴(2%)이었습니다. 용량 감소로 이어진 가장 흔한 유해 반응은 폐렴(0.7%)이었습니다. 환자의 ≥5%에서 용량 중단이 필요했던 유해 반응에는 발열성 호중구 감소증(20%), 호중구 감소증(20%), 폐렴(14%), 패혈증(진균 제외; 11%) 및 혈소판 감소증(10%)이 포함되었습니다. 골수에서 백혈병이 제거된 환자 중 53%에서 절대 호중구 수(ANC) <500/마이크로리터로 인해 용량이 중단되었습니다.
표 15는 VIALE-A에서 확인된 유해 반응을 보여줍니다.
| 유해 반응 | VENCLEXTA + 아자시티딘 (N = 283) |
위약 + 아자시티딘 (N = 144) |
||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 위장관 장애 | ||||
| 메스꺼움 | 44 | 2 | 35 | <1 |
| 설사a | 43 | 5 | 33 | 3 |
| 구토b | 30 | 2 | 23 | <1 |
| 구내염c | 18 | 1 | 13 | 0 |
| 복통d | 18 | <1 | 13 | 0 |
| 혈액 및 림프계 장애 | ||||
| 발열성 호중구 감소증 | 42 | 42 | 19 | 19 |
| 근골격계 및 결합 조직 장애 | ||||
| 근골격계 통증e | 36 | 2 | 28 | 1 |
| 일반적인 장애 및 투여 부위 상태 | ||||
| 피로f | 31 | 6 | 23 | 2 |
| 부종g | 27 | <1 | 19 | 0 |
| 혈관 장애 | ||||
| 출혈h | 27 | 7 | 24 | 3 |
| 저혈압i | 12 | 5 | 8 | 3 |
| 대사 및 영양 장애 | ||||
| 식욕 감소j | 25 | 4 | 17 | <1 |
| 피부 및 피하 조직 장애 | ||||
| 발진k | 25 | 1 | 15 | 0 |
| 감염 및 기생충 감염 | ||||
| 패혈증l (진균성 제외) | 22 | 22 | 16 | 14 |
| 요로 감염m | 16 | 6 | 9 | 6 |
| 호흡기, 흉곽 및 종격 장애 | ||||
| 호흡 곤란n | 18 | 4 | 10 | 2 |
| 신경계 장애 | ||||
| 현기증o | 17 | <1 | 8 | <1 |
| a설사 및 대장염 포함. b구토 및 토혈 포함. c구내염, 구강 궤양, 점막 염증, 구순염, 아프타성 궤양, 설염 및 혀 궤양 포함. d복통, 상복부 통증, 복부 불편감 및 하복부 통증 포함. e관절통, 요통, 사지 통증, 근골격계 통증, 뼈 통증, 근육통, 목 통증, 비 심장성 흉통, 관절염, 근골격계 흉통, 근골격계 경직, 척추 통증 및 근골격계 불편함 포함. f피로 및 무력증 포함. g말초 부종, 부종, 전신 부종, 눈꺼풀 부종, 얼굴 부종, 음경 부종, 안와 주위 부종 및 붓기 포함. h코피, 혈뇨, 결막 출혈, 객혈, 치질 출혈, 잇몸 출혈, 구강 출혈, 두개 내 출혈, 질 출혈, 뇌출혈, 위장관 출혈, 근육 출혈, 피부 출혈, 상부 위장관 출혈, 항문 출혈, 눈 출혈, 출혈성 위염, 출혈, 요로 출혈, 출혈성 질환, 출혈성 뇌졸중, 출혈성 혈관염, 하부 위장관 출혈, 점막 출혈, 음경 출혈, 시술 후 출혈, 직장 출혈, 망막 출혈, 출혈성 쇼크, 연조직 출혈, 경막하 출혈, 혀 출혈, 요도 출혈, 혈관 천자 부위 출혈, 유리체 출혈 및 상처 출혈 포함. i저혈압 및 기립성 저혈압 포함. j식욕 감퇴 및 식욕 부진 포함. k발진, 구진-반점상 발진, 반점상 발진, 약물 발진, 구진성 발진, 농포성 발진, 습진, 홍반성 발진, 가려운 발진, 여드름 모양 피부염, 홍역 모양 발진, 피부염, 건조성 습진, 박탈성 발진 및 혈관 주위 피부염 포함. l패혈증, 대장균 균혈증, 대장균 패혈증, 패혈성 쇼크, 균혈증, 포도상 구균 균혈증, 클렙시엘라 균혈증, 포도상 구균 패혈증, 연쇄상 구균 균혈증, 장구균 균혈증, 클렙시엘라 패혈증, 슈도모나스 균혈증, 슈도모나스 패혈증, 요로 패혈증, 세균성 패혈증, 클로스트리디움 패혈증, 장구균 패혈증, 호중구 감소증 패혈증 및 연쇄상 구균 패혈증 포함. m요로 감염, 대장균 요로 감염, 방광염, 장구균 요로 감염, 세균성 요로 감염, 급성 신우신염 및 슈도모나스 요로 감염 포함. n호흡 곤란, 운동 시 호흡 곤란 및 안정 시 호흡 곤란 포함. o현기증 및 어지럼증 포함. |
||||
다음은 표 15의 기준을 충족하지 않거나 10% 미만인 임상적으로 중요한 부작용(모든 등급)을 나타냅니다.
담즙 및 간장 질환: 담낭염/담석증a (4%)
감염 및 기생충 감염: 폐렴b (33%)
대사 및 영양 장애: 종양 용해 증후군 (1%)
신경계 장애: 두통c (11%)
검사: 체중 감소 (13%).
a급성 담낭염, 담석증, 담낭염, 만성 담낭염 포함.
b폐렴, 폐 감염, 진균성 폐렴, 클렙시엘라 폐렴, 비정형 폐렴, 하부 호흡기 감염, 바이러스성 폐렴, 진균성 하부 호흡기 감염, 헤모필루스 폐렴, 폐렴구균 폐렴, 호흡기 세포 융합 바이러스 폐렴 포함.
c두통 및 긴장성 두통 포함.
표 16은 VIALE-A에서 확인된 실험실 이상을 보여줍니다.
| 실험실 이상 | VENCLEXTA + 아자시티딘 |
위약 + 아자시티딘 |
||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 혈액학 | ||||
| 호중구 감소 | 98 | 98 | 88 | 81 |
| 혈소판 감소 | 94 | 88 | 94 | 80 |
| 림프구 감소 | 91 | 71 | 72 | 39 |
| 헤모글로빈 감소 | 61 | 57 | 56 | 52 |
| 화학 | ||||
| 빌리루빈 증가 | 53 | 7 | 40 | 4 |
| 칼슘 감소 | 51 | 6 | 39 | 9 |
| 나트륨 감소 | 46 | 14 | 47 | 8 |
| 알칼리성 포스파타제 증가 | 42 | 1 | 29 | <1 |
| 혈액 중탄산염 감소 | 31 | <1 | 25 | 0 |
| 최소한 하나의 치료 후 값을 가진 환자 수에 따라 PBO+AZA 군의 경우 분모는 85에서 144까지, VEN+AZA 군의 경우 125에서 283까지 다양했습니다. | ||||
VENCLEXTA와 아자시티딘 또는 데시타빈 병용
신규 진단된 AML 환자를 대상으로 한 비무작위 임상 시험인 M14-358에서 아자시티딘(N=67) 또는 데시타빈(N=13)과 병용한 VENCLEXTA의 안전성을 평가했습니다. 기준선에서 환자는 ≥75세였거나 다음 기준 중 하나 이상에 따라 집중적 유도 화학 요법을 사용할 수 없는 동반 질환이 있었습니다. 기준선 ECOG 수행 상태 2-3, 심각한 심장 또는 폐 동반 질환, 중등도 간 기능 장애, CLcr <45 mL/min 또는 기타 동반 질환 [임상 연구(14.2)]를 참조하십시오. 환자는 램프업 단계 완료 후 아자시티딘(75 mg/m2 정맥 또는 피하 주사, 28일 주기의 1-7일) 또는 데시타빈(20 mg/m2 정맥 주사, 28일 주기의 1-5일)과 병용하여 VENCLEXTA 400 mg을 1일 1회 경구 투여 받았습니다.
아자시티딘
아자시티딘과 병용 투여한 경우 VENCLEXTA에 대한 중간 노출 기간은 6.5개월(범위: 0.1~38.1개월)이었습니다. 이 임상 시험에서 아자시티딘과 병용한 VENCLEXTA의 안전성은 VIALE-A의 안전성과 일치합니다.
데시타빈
데시타빈과 병용 투여한 경우 VENCLEXTA에 대한 중간 노출 기간은 8.4개월(범위: 0.5~39개월)이었습니다.
데시타빈과 함께 VENCLEXTA를 투여받은 환자의 85%에서 중대한 유해 반응이 보고되었으며, 가장 흔한 반응(≥10%)은 패혈증(진균성 제외; 46%), 발열성 호중구 감소증(38%) 및 폐렴(31%)이었습니다. 치료 시작 후 30일 이내에 1명(8%)의 치명적인 유해 반응인 균혈증이 발생했습니다.
유해 반응으로 인해 VENCLEXTA를 영구적으로 중단한 환자는 38%였습니다. 영구적인 중단으로 이어진 가장 흔한 유해 반응(≥5%)은 폐렴(8%)이었습니다.
유해 반응으로 인해 VENCLEXTA의 용량을 감소시킨 환자는 15%였습니다. 용량 감소로 이어진 가장 흔한 유해 반응(≥5%)은 호중구 감소증(15%)이었습니다.
유해 반응으로 인해 VENCLEXTA의 용량을 중단한 환자는 69%였습니다. 용량 중단으로 이어진 가장 흔한 유해 반응(≥10%)은 호중구 감소증(38%), 발열성 호중구 감소증(23%), 백혈구 감소증(15%) 및 폐렴(15%)이었습니다.
가장 흔한 유해 반응(≥30%)은 발열성 호중구 감소증(69%), 피로(62%), 변비(62%), 근골격통(54%), 현기증(54%), 메스꺼움(54%), 복통(46%), 설사(46%), 폐렴(46%), 패혈증(진균성 제외; 46%), 기침(38%), 발열(31%), 저혈압(31%), 인후통(31%), 부종(31%) 및 구토(31%)였습니다. 가장 흔한 실험실 이상(≥30%)은 호중구 감소(100%), 림프구 감소(100%), 백혈구 감소(100%), 혈소판 감소(92%), 칼슘 감소(85%), 헤모글로빈 감소(69%), 글루코스 증가(69%), 마그네슘 감소(54%), 칼륨 감소(46%), 빌리루빈 증가(46%), 알부민 감소(38%), 알칼리성 포스파타제 증가(38%), 나트륨 감소(38%), ALT 증가(31%), 크레아티닌 증가(31%) 및 칼륨 증가(31%)였습니다.
VENCLEXTA와 저용량 시타라빈 병용
VIALE-C
신규 진단된 AML 환자를 대상으로 한 이중맹검 무작위 임상 시험인 VIALE-C에서 저용량 시타라빈(VEN+LDAC)(N=142)과 병용한 VENCLEXTA의 안전성을 위약과 저용량 시타라빈(PBO+LDAC)(N=68)과 비교하여 평가했습니다. 기준선에서 환자는 ≥75세였거나 다음 기준 중 하나 이상에 따라 집중적 유도 화학 요법을 사용할 수 없는 동반 질환이 있었습니다. 기준선 ECOG 수행 상태 2-3, 심각한 심장 또는 폐 동반 질환, 중등도 간 기능 장애, CLcr <45 mL/min 또는 기타 동반 질환 [임상 연구(14.2)]를 참조하십시오. 환자는 4일 램프업 단계 완료 후 저용량 시타라빈(20 mg/m2 피하 주사, 28일 주기의 1-10일)과 병용하여 VENCLEXTA 600 mg을 1일 1회 경구 투여 받거나 위약과 저용량 시타라빈을 투여 받도록 무작위 배정되었습니다. VEN+LDAC를 투여받은 환자 중 VENCLEXTA에 대한 중간 노출 기간은 3.9개월(범위: <0.1~17.1개월)이었습니다.
VEN+LDAC를 투여받은 환자의 65%에서 중대한 유해 반응이 보고되었으며, 가장 흔한 반응(≥10%)은 폐렴(17%), 발열성 호중구 감소증(16%) 및 패혈증(진균성 제외; 12%)이었습니다. VEN+LDAC를 투여받은 환자의 23%에서 치명적인 유해 반응이 발생했으며, 가장 흔한 반응(≥5%)은 폐렴(6%) 및 패혈증(진균성 제외; 7%)이었습니다.
유해 반응으로 인해 VENCLEXTA를 영구적으로 중단한 환자는 25%, 용량을 감소시킨 환자는 9%, 용량을 중단한 환자는 63%였습니다. VENCLEXTA를 영구적으로 중단하게 된 가장 흔한 유해 반응(>2%)은 폐렴(6%)이었습니다. 1% 이상의 환자에서 용량 감소가 필요했던 유해 반응은 폐렴(1%) 및 혈소판 감소증(1%)이었으며, 5% 이상의 환자에서 용량 중단이 필요했던 유해 반응은 호중구 감소증(20%), 혈소판 감소증(15%), 폐렴(8%), 발열성 호중구 감소증(6%) 및 패혈증(진균성 제외; 6%)이었습니다. 골수에서 백혈병이 제거된 환자 중 32%는 ANC <500/microliter로 인해 용량을 중단했습니다.
표 17은 VIALE-C에서 확인된 유해 반응을 보여줍니다.
| 유해 반응 | VENCLEXTA + 저용량 시타라빈 (N = 142) |
위약 + 저용량 시타라빈 (N = 68) |
||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 위장관 장애 | ||||
| 메스꺼움 | 42 | 1 | 31 | 0 |
| 설사 | 28 | 3 | 16 | 0 |
| 구토 | 25 | <1 | 13 | 0 |
| 복통a | 15 | <1 | 9 | 3 |
| 구내염b | 15 | 1 | 6 | 0 |
| 혈액 및 림프계 장애 | ||||
| 발열성 호중구 감소증 | 32 | 32 | 29 | 29 |
| 감염 및 기생충 감염 | ||||
| 폐렴c | 29 | 19 | 21 | 21 |
| 혈관 장애 | ||||
| 출혈d | 27 | 8 | 16 | 1 |
| 저혈압e | 11 | 5 | 4 | 1 |
| 근골격계 및 결합 조직 장애 | ||||
| 근골격계 통증f | 23 | 3 | 18 | 0 |
| 일반적인 장애 및 투여 부위 상태 | ||||
| 피로g | 22 | 2 | 21 | 0 |
| 신경계 장애 | ||||
| 두통 | 11 | 0 | 6 | 0 |
| a복통, 상복부 통증, 복부 불편함 및 하복부 통증을 포함합니다. b구내염, 구강 궤양, 아프타성 궤양, 설염, 점막 염증 및 혀 궤양을 포함합니다. c폐렴, 폐 감염, 하부 호흡기 감염, 진균성 폐렴, 진균성 하부 호흡기 감염, 폐포자충 폐렴, 흡인성 폐렴, 거대 세포 바이러스 폐렴 및 슈도모나스 폐렴을 포함합니다. d코피, 결막 출혈, 객혈, 위장관 출혈, 치은 출혈, 구강 출혈, 상부 위장관 출혈, 혈뇨, 망막 출혈, 카테터 부위 출혈, 뇌출혈, 위 출혈, 출혈성 위염, 두개 내 출혈, 피하 출혈, 입술 출혈, 점막 출혈, 인두 출혈, 시술 후 출혈, 폐포 출혈, 폐 출혈, 치아 펄프 출혈, 자궁 출혈 및 혈관 접근 부위 출혈을 포함합니다. e저혈압 및 기립성 저혈압을 포함합니다. f요통, 관절통, 사지 통증, 근골격계 통증, 근육통, 목 통증, 비 심장성 흉통, 관절염, 뼈 통증, 근골격계 흉통 및 척추 통증을 포함합니다. g피로 및 무력증을 포함합니다. |
||||
다음은 표 17의 기준을 충족하지 않거나 10% 미만인 임상적으로 중요한 부작용(모든 등급)을 나타냅니다.
담즙 및 간 질환: 담낭염/담석증a (1%)
감염 및 기생충 감염: 패혈증b (진균 제외; 15%), 요로 감염c (8%)
대사 및 영양 장애: 식욕 감소 (19%), 종양 용해 증후군 (6%)
신경계 장애: 현기증d (9%)
호흡기, 흉곽 및 종격동 장애: 호흡 곤란e (10%)
검사: 체중 감소 (9%).
a담낭염 및 급성 담낭염 포함.
b패혈증, 균혈증, 패혈성 쇼크, 호중구 감소증 관련 패혈증, 포도상구균 균혈증, 연쇄상구균 균혈증, 세균성 패혈증, 대장균 균혈증, 슈도모나스 균혈증 및 포도상구균 패혈증 포함.
c요로 감염 및 대장균 요로 감염 포함.
d현기증 및 어지럼증 포함.
e호흡 곤란 및 운동 시 호흡 곤란 포함.
표 18은 VIALE-C에서 확인된 실험실 검사 이상을 설명합니다.
| 실험실 검사 이상 | VENCLEXTA + 저용량 시타라빈 |
위약 + 저용량 시타라빈 |
||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 혈액학 | ||||
| 혈소판 감소 | 97 | 95 | 92 | 90 |
| 호중구 감소 | 95 | 92 | 82 | 71 |
| 림프구 감소 | 92 | 69 | 65 | 24 |
| 헤모글로빈 감소 | 63 | 57 | 57 | 54 |
| 화학 | ||||
| 빌리루빈 증가 | 61 | 7 | 38 | 7 |
| 알부민 감소 | 61 | 6 | 43 | 4 |
| 칼륨 감소 | 56 | 16 | 42 | 14 |
| 칼슘 감소 | 53 | 8 | 45 | 13 |
| 포도당 증가 | 52 | 13 | 59 | 9 |
| AST 증가 | 36 | 6 | 37 | 1 |
| 알칼리성 포스파타제 증가 | 34 | 1 | 26 | 1 |
| ALT 증가 | 30 | 4 | 26 | 1 |
| 나트륨 증가 | 11 | 3 | 6 | 1 |
M14-387
M14-387은 새로 진단된 AML 환자를 대상으로 한 비무작위 배정, 공개 표지 시험으로, 저용량 시타라빈(N=61)과 병용한 VENCLEXTA의 안전성을 평가했습니다 [임상 연구(14.2)]. 기준선에서 환자는 ≥75세였거나 다음 기준 중 하나 이상에 따라 집중 유도 화학 요법을 사용할 수 없는 동반 질환이 있었습니다. 기준선 ECOG 수행 상태 2-3, 심각한 심장 또는 폐 동반 질환, 중등도 간 기능 장애, CLcr <45 mL/min 또는 기타 동반 질환. 환자는 램프업 단계 완료 후 저용량 시타라빈(28일 주기의 1-10일째에 피하 20mg/m2)과 병용하여 VENCLEXTA 600mg을 1일 1회 경구 투여 받았습니다. 저용량 시타라빈과 병용한 VENCLEXTA의 안전성은 VIALE-C와 일치합니다.
7 약물 상호작용
7.1 VENCLEXTA에 대한 다른 약물의 영향
강력하거나 중등도의 CYP3A 억제제 또는 P-gp 억제제
강력하거나 중등도의 CYP3A 억제제 또는 P-gp 억제제와의 병용은 베네토클락스 Cmax 및 AUC0-INF를 증가시킵니다 [임상 약리학 (12.3) 참조]. 이는 TLS의 위험을 포함하여 VENCLEXTA의 독성을 증가시킬 수 있습니다 [경고 및 주의 사항 (5.1) 참조].
CLL/SLL 환자에서 초기 및 증량 단계에서 강력한 CYP3A 억제제와의 병용은 금기입니다 [금기 사항 (4) 참조].
CLL/SLL 환자에서 일정한 일일 복용량을 복용하는 경우(증량 단계 이후), 대체 약물을 고려하거나 VENCLEXTA 복용량을 조정하고 부작용을 더 자주 모니터링하십시오 [복용량 및 투여 (2.5, 2.6) 참조].
AML 환자의 경우 VENCLEXTA 복용량을 조정하고 부작용을 더 자주 모니터링하십시오 [복용량 및 투여 (2.5, 2.6) 참조].
강력하거나 중등도의 CYP3A 억제제 또는 P-gp 억제제와의 병용을 중단한 후 2~3일 후에 병용 전에 사용했던 VENCLEXTA 복용량을 재개하십시오 [복용량 및 투여 (2.5, 2.6) 참조].
VENCLEXTA 치료 중에는 자몽, 세비야 오렌지 및 스타후르츠를 피하십시오. 이러한 과일에는 CYP3A 억제제가 포함되어 있습니다.
강력하거나 중등도의 CYP3A 유도제
강력한 CYP3A 유도제와의 병용은 베네토클락스 Cmax 및 AUC0-INF를 감소시킵니다 [임상 약리학 (12.3) 참조]. 이는 VENCLEXTA의 효능을 감소시킬 수 있습니다. 강력한 CYP3A 유도제 또는 중등도의 CYP3A 유도제와 VENCLEXTA를 병용하지 마십시오.
7.2 VENCLEXTA의 다른 약물에 대한 영향
와파린
VENCLEXTA와의 병용은 와파린 Cmax 및 AUC0-INF를 증가시킵니다 [임상 약리학 (12.3) 참조]. 이는 출혈 위험을 증가시킬 수 있습니다. VENCLEXTA와 와파린을 병용하는 환자의 경우 INR을 더 자주 모니터링하십시오.
P-gp 기질
VENCLEXTA와의 병용은 P-gp 기질의 Cmax 및 AUC0-INF를 증가시킵니다 [임상 약리학 (12.3) 참조]. 이는 이러한 기질의 독성을 증가시킬 수 있습니다. VENCLEXTA와 P-gp 기질을 병용하지 마십시오. 병용이 불가피한 경우 VENCLEXTA 복용 전 최소 6시간 간격을 두고 P-gp 기질을 복용하십시오.
8 특정 집단에서의 사용
8.1 임신
위험 요약
동물 연구 결과 및 작용 기전 [임상 약리학 (12.1)]을 기반으로, VENCLEXTA는 임산부에게 투여될 경우 태아에게 해를 끼칠 수 있습니다. VENCLEXTA를 임산부에게 사용한 데이터가 없어 약물 관련 위험을 알 수 없습니다. 임신 중인 쥐에게 오르가노제네시스 기간 동안 베네토클락스를 투여한 결과, AUC 기준으로 일일 400mg의 권장 용량에서 인간 노출량의 1.2배에 해당하는 노출량에서 태아 독성이 나타났습니다. 임산부에게 태아에 대한 잠재적 위험을 알려야 합니다.
지정된 모집단의 주요 선천적 기형 및 유산의 추정 배경 위험은 알려져 있지 않습니다. 모든 임신에는 선천적 기형, 손실 또는 기타 부작용의 배경 위험이 있습니다. 미국 일반 인구에서 임상적으로 인식된 임신에서 주요 선천적 기형 및 유산의 추정 배경 위험은 각각 2%에서 4% 및 15%에서 20%입니다.
데이터
동물 데이터
배아-태아 발달 연구에서 베네토클락스를 오르가노제네시스 기간 동안 임신한 쥐와 토끼에게 투여했습니다. 쥐에서 베네토클락스는 일일 150mg/kg(권장 용량인 일일 400mg에서 인간 노출량의 약 1.2배에 해당하는 모체 노출량)에서 착상 후 손실 증가 및 태아 체중 감소와 관련이 있었습니다. 쥐와 토끼 모두에서 기형 유발성은 관찰되지 않았습니다.
8.2 수유
위험 요약
모유에서 VENCLEXTA의 존재 또는 모유 수유 아기 또는 모유 생산에 미치는 영향에 대한 데이터가 없습니다. 베네토클락스는 수유 쥐에게 투여했을 때 모유에 존재했습니다. (데이터 참조).
모유 수유 아기에게 심각한 부작용이 발생할 가능성이 있으므로, 여성에게 VENCLEXTA 치료 중 및 마지막 투여 후 1주일 동안 모유 수유를 하지 않도록 조언해야 합니다.
데이터
동물 데이터
베네토클락스를 분만 후 8~10일 동안 수유 쥐에게 (단일 용량; 경구 150mg/kg) 투여했습니다. 모유에서 베네토클락스는 혈장보다 1.6배 낮았습니다. 모유에서 총 약물 관련 물질의 대부분은 모 약물(베네토클락스)이었으며, 3가지 대사체가 미량 검출되었습니다.
8.3 생식 능력이 있는 여성 및 남성
VENCLEXTA는 임산부에게 투여될 경우 태아에게 해를 끼칠 수 있습니다. [특정 모집단에서의 사용 (8.1)].
임신 검사
VENCLEXTA 투여를 시작하기 전에 생식 능력이 있는 여성의 임신 여부를 확인하십시오.
피임
생식 능력이 있는 여성에게 VENCLEXTA 치료 중 및 마지막 투여 후 30일 동안 효과적인 피임 방법을 사용하도록 조언하십시오.
불임
동물 연구 결과를 기반으로, VENCLEXTA는 남성의 생식 능력을 손상시킬 수 있습니다. [비임상 독성학 (13.1)].
8.4 소아 사용
소아 환자에서 VENCLEXTA의 안전성 및 유효성은 확립되지 않았습니다.
어린 동물 독성 데이터
어린 동물 독성 연구에서 쥐에게 7일부터 60일까지 경구 투여로 일일 10, 30 또는 100mg/kg의 베네토클락스를 투여했습니다. 독성의 임상 증상으로는 활동 감소, 탈수, 피부 창백 및 구부러진 자세가 ≥30mg/kg/day에서 나타났습니다. 또한 사망률 및 체중 변화는 100mg/kg/day에서 발생했습니다. 베네토클락스와 관련된 다른 영향으로는 ≥10mg/kg/day에서 림프구의 가역적 감소가 있었습니다. 10mg/kg/day의 용량은 20kg 어린이의 경우 400mg의 임상 용량의 약 0.06배에 해당합니다(mg/m2 기준).
8.5 노인 사용
만성 림프구성 백혈병/소림프구성 림프종
VENCLEXTA 단독 요법의 3개의 공개 라벨 시험에서 안전성을 평가한 352명의 이전에 치료받은 CLL/SLL 환자 중 57%(201/352)가 65세 이상이었고 18%(62/352)가 75세 이상이었습니다.
조합 및 단독 요법 연구에서 노인 환자와 젊은 환자 간에 안전성 및 유효성에 임상적으로 의미 있는 차이가 관찰되지 않았습니다.
급성 골수성 백혈병
VIALE-A에서 아자시티딘과 함께 VENCLEXTA를 투여받은 283명의 환자 중 96%가 65세 이상이었고 60%가 75세 이상이었습니다.
M14-358에서 데시타빈과 함께 VENCLEXTA를 투여받은 13명의 환자 중 100%가 65세 이상이었고 62%가 75세 이상이었습니다.
VIALE-C에서 저용량 시타라빈과 함께 VENCLEXTA를 투여받은 142명의 환자 중 92%가 65세 이상이었고 57%가 75세 이상이었습니다.
AML 환자에서 VENCLEXTA의 임상 연구에는 65세 이상 환자가 젊은 성인과 다르게 반응하는지 여부를 확인하기에 충분한 수의 젊은 성인이 포함되지 않았습니다.
8.6 신장애
TLS 위험 증가로 인해, 감소된 신장 기능(Cockcroft-Gault 공식으로 계산한 CLcr <80 mL/min)을 가진 환자는 VENCLEXTA 치료를 시작할 때 TLS 위험을 줄이기 위해 더 집중적인 예방 및 모니터링이 필요합니다 [용법 및 용량 (2.1, 2.2, 2.3, 2.4) 및 경고 및 주의 사항 (5.1)].
경증, 중등도 또는 중증 신장애(CLcr ≥15 mL/min) 환자의 경우 용량 조절은 권장되지 않습니다 [임상 약리학 (12.3)].
8.7 간장애
경증(Child-Pugh A) 또는 중등도(Child-Pugh B) 간장애 환자의 경우 용량 조절은 권장되지 않습니다.
중증 간장애(Child-Pugh C) 환자의 경우 VENCLEXTA 용량을 줄이고, 이러한 환자의 경우 부작용을 더 자주 모니터링해야 합니다 [용법 및 용량 (2.5, 2.7) 및 임상 약리학 (12.3)].
10 과다 복용
VENCLEXTA에 대한 특정 해독제는 없습니다. 과량 복용한 환자의 경우, 면밀히 모니터링하고 적절한 지지 치료를 제공하십시오. 증량 단계 동안 VENCLEXTA를 중단하고 TLS의 징후 및 증상과 기타 독성을 주의 깊게 모니터링하십시오 [용법 및 용량 (2.2, 2.3, 2.4, 2.5)]. 베네토클락스의 광범위한 분포 용적 및 광범위한 단백질 결합을 고려할 때, 투석은 베네토클락스의 유의미한 제거로 이어지지 않을 가능성이 높습니다.
11 설명
베네토클렉스는 BCL-2 억제제입니다. 베네토클렉스는 밝은 노란색에서 어두운 노란색 고체이며, 경험적 공식은 C45H50ClN7O7S이고 분자량은 868.44입니다. 베네토클렉스는 화학적으로 4-(4-{[2-(4-클로로페닐)-4,4-디메틸시클로헥스-1-엔-1-일]메틸}피페라진-1-일)-N-({3-니트로-4-[(테트라히드로-2H-피란-4-일메틸)아미노]페닐}설포닐)-2-(1H-피롤로[2,3-b]피리딘-5-일옥시)벤즈아미드)로 설명되며 다음과 같은 화학 구조를 가지고 있습니다.
![베네토클렉스의 다음 화학 구조는 BCL-2 억제제입니다. 밝은 노란색에서 어두운 노란색 고체이며, 경험적 공식은 C45H50ClN7O7S이고 분자량은 868.44입니다. 베네토클렉스는 화학적으로 4-(4-{[2-(4-클로로페닐)-4,4-디메틸시클로헥스-1-엔-1-일]메틸}피페라진-1-일)-N-({3-니트로-4-[(테트라히드로-2H-피란-4-일메틸)아미노]페닐}설포닐)-2-(1H-피롤로[2,3-b]피리딘-5-일옥시)벤즈아미드)로 설명됩니다.](/images/111/venetoclax-spl-01.jpg)
베네토클렉스는 수용성이 매우 낮습니다.
경구용 VENCLEXTA 정제는 10mg, 50mg 또는 100mg의 베네토클렉스를 활성 성분으로 함유하는 연한 노란색 또는 베이지색 정제로 제공됩니다. 각 정제에는 다음과 같은 비활성 성분도 포함되어 있습니다. 코포비돈, 콜로이드성 실리카, 폴리소르베이트 80, 스테아릴 푸마르산 나트륨, 인산 칼슘 이염기성. 또한, 10mg 및 100mg 코팅 정제에는 다음이 포함됩니다. 황색 산화철, 폴리비닐 알코올, 폴리에틸렌 글리콜, 활석, 이산화 티타늄. 50mg 코팅 정제에는 다음이 포함됩니다. 황색 산화철, 적색 산화철, 흑색 산화철, 폴리비닐 알코올, 활석, 폴리에틸렌 글리콜 및 이산화 티타늄. 각 정제는 한쪽에는 “V”로, 다른 쪽에는 정제 강도에 해당하는 “10”, “50” 또는 “100”으로 각인되어 있습니다.
12 임상약리학
12.1 작용 기전
베네토클락스는 항-세포자멸사 단백질인 BCL-2의 선택적이고 경구적으로 생체 이용 가능한 소분자 억제제입니다. BCL-2의 과발현은 CLL 및 AML 세포에서 입증되었으며, 이는 종양 세포 생존을 매개하고 화학 요법제에 대한 내성과 관련이 있습니다. 베네토클락스는 BCL-2 단백질에 직접 결합하여 BIM과 같은 세포자멸사 유도 단백질을 대체하고 미토콘드리아 외막 투과성을 유발하며 카스파제를 활성화하여 세포자멸사 과정을 회복하는 데 도움이 됩니다. 비임상 연구에서 베네토클락스는 BCL-2를 과발현하는 종양 세포에서 세포독성 활성을 보였습니다.
12.2 약력학
CLL/SLL 환자와 AML 환자에서 임상 연구에서 효능에 대한 노출 반응 분석을 기반으로 약물 노출과 반응 가능성이 높아지는 관계가 관찰되었습니다. 안전성에 대한 노출 반응 분석을 기반으로 AML 환자에서 임상 연구에서 약물 노출과 일부 안전성 사건의 가능성이 높아지는 관계가 관찰되었습니다. 리툭시맙과 병용 투여 시 최대 600mg까지 단독 요법으로 최대 1200mg까지 투여한 CLL/SLL 환자에서는 노출-안전성 관계가 관찰되지 않았습니다.
심장 전기 생리
이전에 치료받은 혈액 악성 종양 환자 176명을 대상으로 한 개방형 단일군 시험에서 하루 1회 최대 1200mg(최대 승인 권장 용량의 2배)의 VENCLEXTA를 다중 투여한 경우 QTc 간격에 대한 영향을 평가했습니다. VENCLEXTA는 QTc 간격에 큰 영향을 미치지 않았습니다(즉, >20ms). 베네토클락스 노출과 QTc 간격 변화 사이에는 관계가 없었습니다.
12.3 약동학
저지방 식사와 함께 하루 1회 400mg을 투여한 후 베네토클락스의 평균(± 표준 편차) 정상 상태 Cmax는 2.1 ± 1.1 mcg/mL이고 AUC0-24h는 32.8 ± 16.9 mcg•h/mL였습니다. 베네토클락스 정상 상태 AUC는 150mg에서 800mg(최대 승인 권장 용량의 0.25배에서 1.33배)의 용량 범위에서 용량에 비례하여 증가했습니다. 베네토클락스의 약동학은 시간이 지남에 따라 변하지 않습니다.
흡수
베네토클락스의 최대 혈장 농도는 여러 번 경구 투여 후 식사 상태에서 5~8시간 후에 도달했습니다.
음식의 영향
저지방 식사(약 512 킬로칼로리, 지방 칼로리 25%, 탄수화물 칼로리 60%, 단백질 칼로리 15%)와 함께 투여하면 베네토클락스 노출이 약 3.4배 증가했고 고지방 식사(약 753 킬로칼로리, 지방 칼로리 55%, 탄수화물 칼로리 28%, 단백질 칼로리 17%)와 함께 투여하면 베네토클락스 노출이 금식 상태에 비해 5.1~5.3배 증가했습니다.
분포
베네토클락스는 1~30 마이크로몰(0.87~26 mcg/mL)의 농도 범위에서 혈장 단백질에 높게 결합되어 혈장에서 결합되지 않은 분율이 <0.01입니다. 혈액-혈장 비율의 평균은 0.57이었습니다. 베네토클락스의 명백한 분포 용적(Vdss/F)은 환자에서 256~321L 범위였습니다.
배설
베네토클락스의 말단 제거 반감기는 약 26시간이었습니다.
대사
베네토클락스는 주로 CYP3A에 의해 in vitro에서 대사됩니다. 혈장에서 확인된 주요 대사체인 M27은 베네토클락스보다 BCL-2에 대한 억제 활성이 적어도 58배 낮으며 in vitro에서 모체 AUC의 80%를 차지했습니다.
배설
건강한 피험자에게 방사성 표지된 [14C]-베네토클락스 200mg을 단일 경구 투여한 후 투여량의 >99.9%가 9일 이내에 대변(변경되지 않은 형태로 21%)에서 회수되었고 <0.1%가 소변에서 회수되었습니다.
특정 인구 집단
연령(19~93세), 성별, 체중, 경증에서 중증 신장애(CLcr 15~89 mL/min, Cockcroft-Gault로 계산), 또는 경증에서 중등도 간 기능 장애(정상 총 빌리루빈 및 아스파르테이트 아미노트랜스퍼라제(AST) > 정상 상한(ULN) 또는 총 빌리루빈 1~3배 ULN)에 따라 베네토클락스의 약동학에서 임상적으로 유의미한 차이가 관찰되지 않았습니다. 말기 신장 질환(CLcr <15 mL/min) 또는 투석이 베네토클락스 약동학에 미치는 영향은 알려져 있지 않습니다.
인종 또는 민족 집단
미국에서 등록된 백인, 흑인 및 아시아 환자에서 베네토클락스의 약동학에서 임상적으로 유의미한 차이가 관찰되지 않았습니다. AML 환자 771명 중 아시아 국가 출신의 아시아 환자[중국(5.6%), 일본(5.5%), 한국(2.1%), 대만(0.9%)]는 비아시아 인구보다 베네토클락스 노출이 63% 높았습니다.
간 기능 장애 환자
VENCLEXTA 50mg을 단일 투여한 후 베네토클락스 전신 노출(AUC0-INF)은 중증 간 기능 장애(Child-Pugh C) 환자에서 정상 간 기능 환자에 비해 2.7배 높았습니다. [용량 및 투여(2.7) 및 특정 인구 집단에서의 사용(8.7) 참조]. 경증 또는 중등도 간 기능 장애 환자와 정상 간 기능 환자 사이에서 베네토클락스 전신 노출의 임상적으로 유의미한 차이가 관찰되지 않았습니다.
약물 상호 작용 연구
임상 연구
아자시티딘, 아지트로마이신, 시타라빈, 데시타빈, 위산 감소제, 오비누투주맙 또는 리툭시맙과 병용 투여했을 때 베네토클락스의 약동학에서 임상적으로 유의미한 차이가 관찰되지 않았습니다.
케토코나졸
케토코나졸(강력한 CYP3A, P-gp 및 BCRP 억제제)을 1일 1회 400mg씩 7일 동안 병용 투여하면 베네토클락스 Cmax가 130% 증가하고 AUC0-INF가 540% 증가합니다. [약물 상호 작용(7.1)]를 참조하십시오.
리토나비르
리토나비르(강력한 CYP3A, P-gp 및 OATP1B1/B3 억제제)를 1일 1회 50mg씩 14일 동안 병용 투여하면 베네토클락스 Cmax가 140% 증가하고 AUC가 690% 증가합니다. [약물 상호 작용(7.1)]를 참조하십시오.
포사코나졸
포사코나졸(강력한 CYP3A 및 P-gp 억제제)을 VENCLEXTA 50mg 및 100mg과 함께 7일 동안 300mg 병용 투여하면 베네토클락스 Cmax가 각각 61% 및 86% 증가했습니다. 베네토클락스 AUC0-24h는 각각 90% 및 144% 증가했습니다. [약물 상호 작용(7.1)]를 참조하십시오.
리팜핀
리팜핀 (OATP1B1/1B3 및 P-gp 억제제)을 600mg 단일 용량으로 병용 투여하면 베네토클락스 Cmax가 106% 증가하고 AUC0-INF가 78% 증가했습니다. 리팜핀(강력한 CYP3A 유도제)을 1일 1회 600mg씩 13일 동안 다중 용량으로 병용 투여하면 베네토클락스 Cmax가 42% 감소하고 AUC0-INF가 71% 감소했습니다. [약물 상호 작용(7.1)]를 참조하십시오.
와파린
VENCLEXTA 400mg 단일 용량을 와파린 5mg과 병용 투여하면 R-와파린 및 S-와파린의 Cmax 및 AUC0-INF가 18%에서 28% 증가했습니다. [약물 상호 작용(7.2)]를 참조하십시오.
디곡신
VENCLEXTA 100mg 단일 용량을 디곡신(P-gp 기질) 0.5mg과 병용 투여하면 디곡신 Cmax가 35% 증가하고 AUC0-INF가 9% 증가했습니다. [약물 상호 작용(7.2)]를 참조하십시오.
시험관 내 연구
베네토클락스는 CYP1A2, CYP2B6, CYP2C19, CYP2D6 또는 CYP3A4의 억제제 또는 유도제가 아닙니다. 베네토클락스는 CYP2C8, CYP2C9 및 UGT1A1의 약한 억제제입니다.
베네토클락스는 UGT1A4, UGT1A6, UGT1A9 또는 UGT2B7의 억제제가 아닙니다.
베네토클락스는 P-gp 및 BCRP의 억제제이자 기질이며 OATP1B1의 약한 억제제입니다.
베네토클락스는 OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1 또는 MATE2K의 억제제가 아닙니다.
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 저해
베네토클렉스 또는 주요 인체 대사체인 M27은 6개월 동안 경구 투여량이 최대 400 mg/kg/일인 베네토클렉스와 단일 경구 투여량이 250 mg/kg/일인 M27에서 형질전환 (Tg.rasH2) 마우스 연구에서 발암성이 없었습니다.
베네토클렉스는 시험관 내 박테리아 돌연변이 유발성 (Ames) 검사에서 돌연변이 유발성이 없었고, 인간 말초 혈액 림프구를 사용한 시험관 내 염색체 이상 검사에서 수적 또는 구조적 이상을 유발하지 않았으며, 최대 835 mg/kg의 투여량에서 시험관 내 마우스 골수 미세핵 검사에서 클라스토젠성이 없었습니다. M27 대사체는 시험관 내 Ames 및 염색체 이상 검사에서 유전 독성 활성이 음성이었습니다.
수컷 및 암컷 마우스에서 생식력 및 초기 배아 발달 연구가 수행되었습니다. 이러한 연구는 교미, 수정 및 착상을 통한 배아 발달을 평가합니다. 최대 600 mg/kg/일의 투여량에서 발정 주기, 교미, 생식력, 황체, 자궁 착상 또는 새끼당 살아있는 배아에 대한 베네토클렉스의 영향은 없었습니다. 그러나 400 mg의 투여량에서 인간 AUC 노출의 0.5배만큼 낮은 노출에서 개에서 관찰된 고환 독성 (생식 세포 손실)을 기반으로 인간 남성 생식력에 대한 위험이 존재합니다.
13.2 동물 독성 및/또는 약리학
개에서 베네토클렉스는 조직 무결성 또는 기관 기능 장애의 증거 없이 담낭, 외분비 췌장 및 위를 포함한 다양한 조직에서 단일 세포 괴사를 유발했습니다. 이러한 발견은 규모가 최소에서 경미했습니다. 4주간의 투약 기간과 그 후 4주간의 회복 기간 후, 일부 조직에서 최소한의 단일 세포 괴사가 여전히 존재했으며, 더 긴 투약 또는 회복 기간 후에 가역성이 평가되지 않았습니다.
또한 개에서 매일 투약 후 약 3개월 후, 베네토클렉스는 멜라닌 색소의 손실로 인해 털이 점차 흰색으로 변색되었습니다.
14 임상 연구
14.1 만성 림프구성 백혈병/소림프구성 림프종
오비누투주맙과 병용
CLL14(BO25323)는 이전에 치료를 받지 않은 CLL 환자로서 동반 질환(총 누적 질병 평가 척도 [CIRS] 점수 >6 또는 CLcr <70 mL/min)이 있는 환자를 대상으로 한 무작위 배정(1:1), 다기관, 공개, 적극적 대조군 시험(NCT02242942)으로, 오비누투주맙과 병용한 VENCLEXTA(VEN+G)의 효능과 안전성을 오비누투주맙과 병용한 클로람부실(GClb)과 비교 평가했습니다. 이 시험에서는 간 트랜스아미나제와 총 빌리루빈이 정상 상한치의 2배 이하이어야 했으며, 리히터 변형 또는 CIRS에 의한 눈, 귀, 코, 인후를 제외한 모든 개별 기관/시스템 장애 점수가 4인 환자는 제외했습니다.
모든 환자는 1주기 1일(첫 번째 투여는 1일 100mg과 2일 900mg으로 나눌 수 있음), 8일, 15일에 1000mg의 오비누투주맙을 투여받았으며, 이후 각 주기의 1일에 총 6주기 동안 투여받았습니다. VEN+G군 환자는 1주기 22일에 VENCLEXTA 5주 증량 투여 일정 [투여 및 관리(2.2, 2.4)]을 시작했으며, 3주기 1일부터 12주기 마지막 날까지 매일 1회 경구로 VENCLEXTA 400mg을 투여받았습니다. GClb군에 무작위 배정된 환자는 1주기부터 12주기까지 1일과 15일에 경구로 클로람부실 0.5mg/kg을 투여받았습니다. 각 주기는 28일이었습니다.
총 432명의 환자가 무작위 배정되었으며, 각 군에 216명이 배정되었습니다. 기준선 인구 통계학적 특징과 질병 특징은 군 간에 유사했습니다. 중앙값 연령은 72세(범위: 41~89세)였으며, 89%가 백인, 67%가 남성이었습니다. 36%와 43%가 각각 비네 단계 B와 C였으며, 88%가 동부 협력 종양학 그룹(ECOG) 수행 상태 <2였습니다. 중앙값 CIRS 점수는 8.0(범위: 0~28)이었으며, 58%의 환자가 CLcr <70 mL/min이었습니다. 17p 결손은 8%의 환자에서, TP53 돌연변이는 10%에서, 11q 결손은 19%에서, 돌연변이가 없는 IgVH는 57%에서 발견되었습니다.
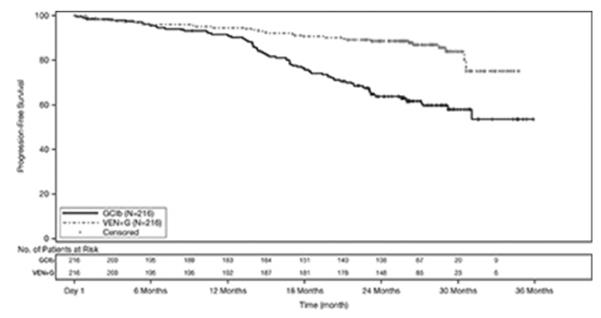
효능은 독립적인 검토 위원회(IRC)가 평가한 무진행 생존율(PFS)을 기반으로 했습니다. PFS에 대한 중앙값 추적 기간은 28개월(범위: 0~36개월)이었습니다. CLL14에 대한 효능 결과는 표 19에 나와 있습니다. PFS에 대한 Kaplan-Meier 곡선은 그림 1에 나와 있습니다.
| 종점 | VENCLEXTA + 오비누투주맙 (N = 216) |
오비누투주맙 + 클로람부실 (N = 216) |
| 무진행 생존율a | ||
| 사건 수, n (%) | 29 (13) | 79 (37) |
| 질병 진행 | 14 (6) | 71 (33) |
| 사망 | 15 (7) | 8 (4) |
| 중앙값, 개월 | 달성되지 않음 | 달성되지 않음 |
| HR (95% CI)b | 0.33 (0.22, 0.51) | |
| p-값b | <0.0001 | |
| 반응률c, n (%) | ||
| ORRd | 183 (85) | 154 (71) |
| 95% CI | (79, 89) | (65, 77) |
| CR | 100 (46) | 47 (22) |
| CR+CRid | 107 (50) | 50 (23) |
| PR | 76 (35) | 104 (48) |
| CI = 신뢰 구간; CR = 완전 관해; CRi = 불완전 골수 회복을 동반한 완전 관해; HR = 위험 비; ORR = 전반적인 반응률(CR + CRi + PR); PR = 부분 관해. a무작위 배정부터 질병 진행 또는 사망(원인 불문)의 가장 빠른 사건까지. IRC 평가; Kaplan-Meier 추정. bHR 추정치는 비네 단계와 지리적 지역에 따라 계층화된 Cox 비례 위험 모델을 기반으로 합니다. p-값은 동일한 요인에 따라 계층화된 로그 순위 검정을 기반으로 합니다. c2008년 만성 림프구성 백혈병 국제 워크숍(IWCLL) 지침에 따름. dCochran-Mantel-Haenszel 검정을 기반으로 한 p-값; ORR에 대한 p=0.0007; CR+CRi에 대한 p<0.0001. |
||
그림 1. CLL14에서 IRC 평가 무진행 생존의 Kaplan-Meier 곡선
분석 시점에서 중간 전체 생존율(OS)은 도달하지 않았으며, 10% 미만의 환자가 사건을 경험했습니다. OS에 대한 중간 추적 기간은 28개월이었습니다.
최소 잔여 질환(MRD)은 대립 유전자 특이적 올리고뉴클레오티드 중합효소 연쇄 반응(ASO-PCR)을 사용하여 평가했습니다. 음성 상태의 정의는 104개의 백혈구당 CLL 세포가 1개 미만이었습니다. 치료 완료 후 3개월 후 반응과 관계없이 MRD 음성률과 CR을 달성한 환자의 MRD 음성률은 표 20에 나와 있습니다. 이 평가에서 VEN+G군에서 말초 혈액에서 MRD 음성인 134명의 환자는 일치하는 골수 검체를 보유하고 있었으며, 이 중 122명의 환자(91%)는 말초 혈액과 골수 모두에서 MRD 음성이었습니다.
| VENCLEXTA + 오비누투주맙 |
오비누투주맙 + 클로람부실 |
|
| MRD 음성률(ITT 모집단) | ||
| N | 216 | 216 |
| 골수, n (%) | 123 (57) | 37 (17) |
| 95% CI | (50, 64) | (12, 23) |
| p-값a | <0.0001 | |
| 말초 혈액, n (%) | 163 (76) | 76 (35) |
| 95% CI | (69, 81) | (29, 42) |
| p-값a | <0.0001 | |
| CR 환자의 MRD 음성률 | ||
| N | 100 | 47 |
| 골수, n (%) | 69 (69) | 21 (45) |
| 95% CI | (59, 78) | (30, 60) |
| p-값a | 0.0048 | |
| 말초 혈액, n (%) | 87 (87) | 29 (62) |
| 95% CI | (79, 93) | (46, 75) |
| p-값a | 0.0005 | |
| CI = 신뢰 구간; CR = 완전 관해. a카이제곱 검정 기반 p-값. |
||
치료 완료 후 12개월 시점에서 말초혈액에서 MRD 음성률은 VEN+G 치료군에서 58%(126/216)였고, GClb 치료군에서 9%(20/216)였습니다.
리툭시맙 병용
MURANO는 최소 1회 이상의 사전 치료를 받은 CLL 환자에서 리툭시맙 병용 VENCLEXTA(VEN+R)의 효능 및 안전성을 벤다무스틴 병용 리툭시맙(B+R)과 비교 평가한 무작위 배정(1:1), 다기관, 공개 표지 시험(NCT02005471)이었습니다. VEN+R 군 환자는 VENCLEXTA 5주 증량 투약 일정을 완료했습니다 [용법 및 용량(2.2, 2.4)] 그리고 질병 진행 또는 용납할 수 없는 독성이 없는 경우 리툭시맙 1주기 1일부터 24개월 동안 VENCLEXTA 400mg을 1일 1회 경구 투여 받았습니다. 리툭시맙은 5주 증량 투약 후 1주기 1일에 375mg/m2를 정맥 주사하고, 2-6주기 1일에 500mg/m2를 정맥 주사하는 용량으로 시작했습니다. B+R 군에 무작위 배정된 환자는 위에 설명된 용량 및 일정으로 리툭시맙과 병용하여 6주기 동안 1일 및 2일에 벤다무스틴 70mg/m2를 정맥 주사했습니다. 각 주기는 28일이었습니다.
총 389명의 환자가 무작위 배정되었습니다: VEN+R 군 194명, B+R 군 195명. 기준선 인구 통계적 특징 및 질병 특징은 VEN+R 군과 B+R 군에서 유사했습니다. 중앙값 연령은 65세(범위: 22~85세)였고, 97%가 백인, 74%가 남성, 99%가 ECOG 수행 상태 <2였습니다. 사전 치료의 중앙값은 1회(범위: 1~5회)였습니다. 59%는 1회 사전 치료를 받았고, 26%는 2회 사전 치료를 받았고, 16%는 3회 이상의 사전 치료를 받았습니다. 사전 치료에는 알킬화제(94%), 항-CD20 항체(77%), B 세포 수용체 경로 억제제(2%), 사전 퓨린 유사체(81%, 플루다라빈/사이클로포스파미드/리툭시맙 포함 55%)가 포함되었습니다. 17p 결손은 환자의 24%에서, TP53 돌연변이는 25%에서, 11q 결손은 32%에서, 돌연변이가 없는 IgVH는 63%에서 발견되었습니다.
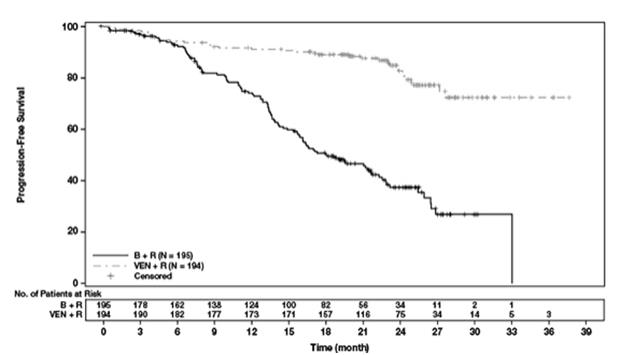
효능은 IRC에 의해 평가된 PFS를 기반으로 했습니다. PFS에 대한 중앙값 추적 관찰 기간은 23.4개월(범위: 0~37.4+개월)이었습니다. MURANO의 효능 결과는 표 21에 나와 있습니다. PFS에 대한 Kaplan-Meier 곡선은 그림 2에 나와 있습니다.
| 종점 | VENCLEXTA + 리툭시맙 (N = 194) |
벤다무스틴 + 리툭시맙 (N = 195) |
| 무진행 생존a | ||
| 사건 수, n (%) | 35 (18) | 106 (54) |
| 질병 진행, n | 26 | 91 |
| 사망 사건, n | 9 | 15 |
| 중앙값, 개월(95% CI) | 도달하지 않음 | 18.1 (15.8, 22.3) |
| HR (95% CI)b | 0.19 (0.13, 0.28) | |
| p-값b | <0.0001 | |
| 반응률c, n (%) | ||
| ORR | 179 (92) | 141 (72) |
| 95% CI | (88, 96) | (65, 78) |
| CR+CRi | 16 (8) | 7 (4) |
| nPR | 3 (2) | 1 (1) |
| PR | 160 (82) | 133 (68) |
| CI = 신뢰 구간; CR = 완전 관해; CRi = 불완전 골수 회복을 동반한 완전 관해; HR = 위험 비; nPR = 결절성 부분 관해; ORR = 전반적인 반응률(CR + CRi + nPR + PR); PR = 부분 관해. aKaplan-Meier 추정치. bHR 추정치는 17p 결손, 위험 상태 및 지리적 지역에 따라 계층화된 Cox 비례 위험 모델을 기반으로 합니다. p-값은 동일한 요인에 따라 계층화된 로그 순위 검정을 기반으로 합니다. c2008년 만성 림프구성 백혈병(IWCLL) 국제 워크숍 지침에 따름. |
||
그림 2. MURANO에서 IRC 평가 무진행 생존의 Kaplan-Meier 곡선
리툭시맙의 마지막 투여 후 3개월에, PR 이상을 달성한 환자의 말초혈액에서 MRD 음성률은 VEN+R 군에서 54%(104/194)였고, B+R 군에서 12%(23/195)였습니다. 이 시점에서 MRD 음성 CR/CRi 비율은 VEN+R 군에서 3%(6/194)였고, B+R 군에서 2%(3/195)였습니다.
71개월 추적 관찰
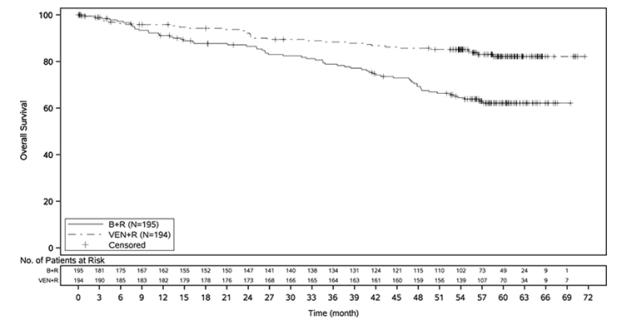
전반적인 추적 관찰 기간이 71개월인 경우, 연구자 평가 중간 PFS는 VEN+R 군에서 53.6개월(95% CI: 48.4, 57.0)이었고, B+R 군에서 17.0개월(95% CI: 15.5, 21.7)이었습니다. 어느 군에서도 중간 OS에 도달하지 못했습니다. VEN+R 군 환자의 16%(32/194)와 B+R 군 환자의 33%(64/195)에서 사망이 발생했습니다(층화된 HR 0.40; 95% CI [0.26, 0.62]). OS에 대한 Kaplan-Meier 곡선은 그림 3에 나와 있습니다.
그림 3. MURANO에서 전체 생존의 Kaplan-Meier 곡선
단독 요법
이전에 치료받은 CLL 또는 SLL에서 VENCLEXTA 단독 요법의 효능은 3개의 단일군 시험을 기반으로 합니다.
M13-982
M13-982(NCT01889186)는 이전에 최소한 1회 이상 치료를 받은 17p 결손이 있는 CLL 환자 106명을 등록한 공개, 다기관 시험이었습니다. 이 시험에서, 17p 결손은 VENCLEXTA 치료를 위한 환자 선택에 FDA 승인을 받은 Vysis CLL FISH Probe Kit를 사용하여 환자의 말초혈액 표본에서 확인되었습니다. 환자는 증량 투여 일정을 완료한 후 VENCLEXTA 400mg을 1일 1회 경구 투여 받았습니다 [투여 및 투약 방법(2.2, 2.4)].
효능은 IRC가 평가한 전체 반응률(ORR)을 기반으로 했습니다.
표 22는 시험 모집단의 기준 인구 통계 및 질병 특성을 요약합니다.
| 특성 | N = 106 |
| 나이, 년; 중간값(범위) | 67 (37-83) |
| 백인; % | 97 |
| 남성; % | 65 |
| ECOG 수행 상태; % 0 1 2 |
40 52 8 |
| 종양 부담; % 절대 림프구 수 ≥25 x 109/L 5cm 이상인 결절 1개 이상 |
50 53 |
| 이전 치료 횟수; 중간값(범위) | 2.5 (1-10) |
| 진단 이후 시간, 년; 중간값(범위)a | 6.6 (0.1-32.1) |
| ECOG = 동부 협력 종양학 그룹. aN=105. |
|
평가 시점의 치료 기간 중간값은 12.1개월(범위: 0~21.5개월)이었습니다. 효능 결과는 표 23에 나와 있습니다.
| 종점 | VENCLEXTA N = 106 |
| ORR, n (%)a (95% CI) |
85 (80) (71, 87) |
| CR + CRi, n (%) CR, n (%) CRi, n (%) |
8 (8) 6 (6) 2 (2) |
| nPR, n (%) | 3 (3) |
| PR, n (%) | 74 (70) |
| CI = 신뢰 구간; CR = 완전 관해; CRi = 불완전 골수 회복을 동반한 완전 관해; IRC = 독립적인 검토 위원회; nPR = 결절성 부분 관해; ORR = 전체 반응률(CR + CRi + nPR + PR); PR = 부분 관해. a2008 IWCLL 지침에 따름. |
|
첫 반응까지의 중간 시간은 0.8개월(범위: 0.1~8.1개월)이었습니다.
나중에 데이터 마감일과 연구자 평가 효능을 기반으로 반응 지속 기간(DOR)은 2.9~32.8+개월이었습니다. 중간 추적 관찰 기간이 22개월인 경우 중간 DOR에 도달하지 않았습니다.
VENCLEXTA 치료 후 CR 또는 CRi를 달성한 환자의 말초 혈액과 골수에서 최소 잔여 질환을 평가했습니다. 3%(3/106)가 말초 혈액과 골수에서 MRD 음성(104 백혈구당 CLL 세포 1개 미만)을 달성했습니다.
M12-175
M12-175(NCT01328626)는 17p 결손이 있는 환자를 포함하여 이전에 치료받은 CLL 또는 SLL 환자를 등록한 개방형 다기관 시험이었습니다. 효능은 VENCLEXTA 400mg을 경구로 1일 1회, 증량 투약 일정을 완료한 후에 받은 67명의 환자(CLL 59명, SLL 8명)에서 평가했습니다. 환자는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 이 용량을 계속했습니다. 평가 당시 치료의 중간 기간은 22.1개월(범위: 0.5~71.7개월)이었습니다.
중간 연령은 65세(범위: 42~84세)였으며, 78%가 남성이었고 87%가 백인이었습니다. 이전 치료의 중간 횟수는 3회(범위: 1~11회)였습니다. 기준선에서 환자의 67%는 5cm 이상의 결절이 1개 이상 있었고, 30%는 ALC가 25 x 109/L 이상이었고, 33%는 변이되지 않은 IgVH가 문서화되었으며, 21%는 17p 결손이 문서화되었습니다.
효능은 2008년 IWCLL 지침을 기반으로 했으며 IRC가 평가했습니다. ORR은 76%(95% CI: 64%, 86%)였으며, CR + CRi 비율은 10%, PR 비율은 66%였습니다. 중간 DOR은 36.2개월(범위: 2.4~52.4개월)이었습니다.
M14-032
M14-032(NCT02141282)는 이전에 이브루티닙 또는 이델랄리십으로 치료받았고 이브루티닙 또는 이델랄리십 치료 후 진행된 CLL 환자를 등록한 개방형 다기관 시험이었습니다. 환자는 증량 투약 일정을 완료한 후 VENCLEXTA 400mg을 경구로 1일 1회 받았습니다 [용량 및 투여(2.2, 2.4)]. 환자는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 이 용량을 계속했습니다. 분석 당시 치료의 중간 기간은 19.5개월(범위: 0.1~39.5개월)이었습니다.
치료받은 127명의 환자(이전에 이브루티닙으로 치료받은 91명, 이전에 이델랄리십으로 치료받은 36명) 중 중간 연령은 66세(범위: 28~85세)였으며, 70%가 남성이었고 92%가 백인이었습니다. 이전 치료의 중간 횟수는 4회(범위: 1~15회)였습니다. 기준선에서 환자의 41%는 5cm 이상의 결절이 1개 이상 있었고, 31%는 ALC가 25 x 109/L 이상이었고, 57%는 변이되지 않은 IgVH가 문서화되었으며, 39%는 17p 결손이 문서화되었습니다.
효능은 2008년 IWCLL 지침을 기반으로 했으며 IRC가 평가했습니다. ORR은 70%(95% CI: 61%, 78%)였으며, CR + CRi 비율은 5%, PR 비율은 65%였습니다. 중간 추적 관찰 기간이 19.9개월(범위: 2.9~36개월)인 경우 중간 DOR에 도달하지 않았습니다.
14.2 급성 골수성 백혈병
VENCLEXTA는 새로 진단된 AML 환자 중 75세 이상이거나 다음 기준 중 하나 이상에 따라 집중 유도 화학 요법을 사용할 수 없는 동반 질환이 있는 성인 환자에서 연구되었습니다. 기준선 ECOG 수행 상태 2-3, 심각한 심장 또는 폐 동반 질환, 중등도 간 기능 장애, CLcr <45 mL/min 또는 기타 동반 질환.
아자시티딘 또는 데시타빈과 병용
VIALE-A는 아자시티딘과 병용한 VENCLEXTA(VEN+AZA)의 효능과 안전성을 위약과 아자시티딘(PBO+AZA)과 비교 평가한 무작위 배정(2:1), 이중 맹검, 위약 대조, 다기관 시험(NCT02993523)이었습니다.
환자는 증량 투약 일정을 완료한 후 1일 1회 경구로 VENCLEXTA 400mg을 받았습니다 [용량 및 투여(2.3)] 또는 위약을 아자시티딘 75mg/m2와 병용하여 1주기 1일 1일에 시작하여 28일 주기의 1~7일에 정맥 또는 피하로 투여했습니다. 증량 기간 동안 환자는 TLS 예방을 받았고 모니터링을 위해 입원했습니다.
골수 평가를 통해 1주기 치료 후 5% 미만의 백혈병 폭발이 있고 혈소판 감소증이 있는 경우 완화, 즉 1주기 치료 후 5% 미만의 백혈병 폭발이 있고 혈소판 감소증이 있는 경우 VENCLEXTA 또는 위약을 최대 14일까지 중단하거나 ANC ≥500/마이크로리터 및 혈소판 수 ≥50 x 103/마이크로리터가 될 때까지 중단했습니다. 1주기 말에 내성 질환이 있는 환자의 경우 2주기 또는 3주기 후에 골수 평가를 수행했고 임상적으로 필요에 따라 수행했습니다. VENCLEXTA 또는 위약 중단 후 같은 날에 아자시티딘을 재개했습니다. 아자시티딘 용량 감소는 혈액학적 독성 관리를 위해 임상 시험에서 시행되었습니다 [용량 및 투여(2.5)]. 환자는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 치료를 계속했습니다.
총 431명의 환자가 무작위 배정되었습니다. VEN+AZA 군 286명, PBO+AZA 군 145명이었습니다. 기준선 인구 통계 및 질병 특징은 표 24에 나와 있습니다.
| 특징 | VENCLEXTA + 아자시티딘 N = 286 |
위약 + 아자시티딘 N = 145 |
| 연령, 년; 중간값(범위) | 76(49, 91) | 76(60, 90) |
| 인종 | ||
| 백인; % | 76 | 75 |
| 흑인 또는 아프리카계 미국인; % | 1 | 1.4 |
| 아시아인; % | 23 | 23 |
| 남성; % | 60 | 60 |
| ECOG 수행 상태; % | ||
| 0-1 | 55 | 56 |
| 2 | 40 | 41 |
| 3 | 5.6 | 3.4 |
| 골수 폭발; % | ||
| <30% | 30 | 28 |
| ≥30% to <50% | 21 | 23 |
| ≥50% | 49 | 49 |
| 질병 이력; % | ||
| 신규 AML | 75 | 76 |
| 이차성 AML | 25 | 24 |
| 검출된 세포 유전학적 위험a, % | ||
| 중간 | 64 | 61 |
| 불량 | 36 | 39 |
| 검출된 돌연변이 분석; n/Nb (%) | ||
| IDH1 또는 IDH2 | 61/245 (25) | 28/127 (22) |
| IDH1 | 23/245 (9.4) | 11/127 (8.7) |
| IDH2 | 40/245 (16) | 18/127 (14) |
| FLT3 | 29/206 (14) | 22/108 (20) |
| NPM1 | 27/163 (17) | 17/86 (20) |
| TP53 | 38/163 (23) | 14/86 (16) |
| a2016년 국립 종합 암 네트워크(NCCN) 가이드라인에 따름. b기준선에서 수령한 평가 가능한 BMA 샘플 수. |
||
효능은 무작위 배정일로부터 모든 원인으로 인한 사망까지 측정된 전체 생존율(OS)을 기반으로 했습니다. VEN+AZA의 조합은 PBO+AZA보다 OS에서 우수했습니다.
OS에 대한 Kaplan-Meier 곡선은 그림 4에 나와 있습니다. VIALE-A의 효능 결과는 표 25에 나와 있습니다.
그림 4. VIALE-A에서 전체 생존율에 대한 Kaplan-Meier 곡선
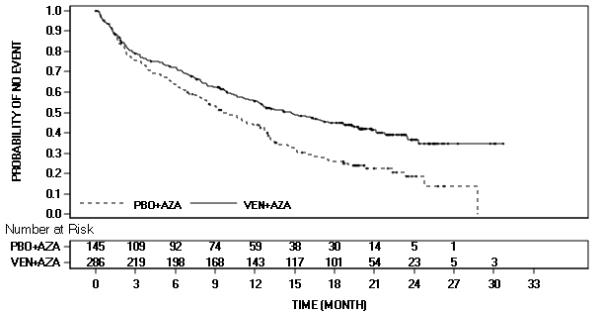
| 종점 | VENCLEXTA + 아자시티딘 (N = 286) |
위약 + 아자시티딘 (N = 145) |
| 전체 생존율 | ||
| 중앙값a, 개월 (95% CI) |
14.7 (11.9, 18.7) |
9.6 (7.4, 12.7) |
| 위험 비율b (95% CI) | 0.66 (0.52, 0.85) | |
| p-값b | <0.001 | |
| 반응률 | ||
| CR, n (%) | 105 (37) | 26 (18) |
| (95% CI) | (31, 43) | (12, 25) |
| p-값c | <0.001 | |
| DOCR 중앙값a,d (개월) | 18.0 | 13.4 |
| 95% CI | (15.3, -) | (8.7, 17.6) |
| CR+CRh, n (%) | 185 (65) | 33 (23) |
| (95% CI) | (59, 70) | (16, 30) |
| p-값c | <0.001 | |
| DOCR+CRh 중앙값a,e (개월) | 17.8 | 13.9 |
| 95% CI | (15.3, -) | (10.4, 15.7) |
| CI = 신뢰 구간; CR = 완전 관해; CRh = 부분 혈액학적 회복을 동반한 완전 관해; DOCR = CR 기간; HR = 위험 비율; – = 도달하지 않음. CR(완전 관해)는 절대 호중구 수 >1,000/마이크로리터, 혈소판 >100,000/마이크로리터, 적혈구 수혈 독립성 및 <5% 폭발이 있는 골수로 정의되었습니다. 순환 폭발 및 Auer 막대가 있는 폭발의 부재; 골수 외 질환의 부재. CRh(부분 혈액학적 회복을 동반한 완전 관해)는 골수에서 <5%의 폭발, 질병의 증거가 없고 말초 혈액 수치의 부분적 회복(혈소판 >50,000/마이크로리터 및 ANC >500/마이크로리터)으로 정의되었습니다. aKaplan-Meier 추정치. b위험 비율 추정치(VEN+AZA 대 PBO+AZA)는 무작위 배정 시 할당된 염색체 이상(중간 위험, 불량 위험) 및 연령(18세에서 <75세, ≥75세)으로 계층화된 Cox 비례 위험 모델을 기반으로 합니다. p-값은 동일한 요인으로 계층화된 로그 랭크 검정을 기반으로 합니다. cp-값은 연령 및 염색체 이상 위험으로 계층화된 Cochran-Mantel-Haenszel 검정의 결과입니다. dCR 기간은 첫 번째 CR 반응 날짜부터 확인된 형태학적 재발, 확인된 진행성 질환 또는 질병 진행으로 인한 사망의 가장 빠른 증거 날짜까지의 일수로 정의됩니다. eCR+CRh 기간은 첫 번째 CR+CRh(CR 또는 CRh 중 첫 번째) 반응 날짜부터 확인된 형태학적 재발, 확인된 진행성 질환 또는 질병 진행으로 인한 사망의 가장 빠른 증거 날짜까지의 일수로 정의됩니다. |
||
VEN+AZA로 치료받은 환자 중 155명은 기준선에서 적혈구(RBC) 및/또는 혈소판 수혈에 의존적이었으며, 이러한 환자 중 49%(76/155)는 기준선 이후 연속적인 ≥56일 동안 RBC 및 혈소판 수혈에서 독립적으로 되었습니다. VEN+AZA로 치료받은 환자 중 131명은 기준선에서 RBC 및 혈소판 수혈 모두에 독립적이었으며, 69%(90/131)는 기준선 이후 연속적인 ≥56일 동안 수혈 독립성을 유지했습니다. PBO+AZA로 치료받은 환자 중 81명은 기준선에서 RBC 및/또는 혈소판 수혈에 의존적이었으며, 이러한 환자 중 27%(22/81)는 기준선 이후 연속적인 ≥56일 동안 RBC 및 혈소판 수혈에서 독립적으로 되었습니다. PBO+AZA로 치료받은 환자 중 64명은 기준선에서 RBC 및 혈소판 수혈 모두에 독립적이었으며, 42%(27/64)는 기준선 이후 연속적인 ≥56일 동안 수혈 독립성을 유지했습니다.
VEN+AZA 치료를 받은 환자의 CR 또는 CRh에 대한 최초 반응까지의 중앙값은 1.0개월(범위: 0.6~14.3개월)이었습니다.
M14-358
M14-358(NCT02203773)은 새로 진단된 AML 환자에서 아자시티딘(N=84) 또는 데시타빈(N=31)과 함께 VENCLEXTA의 효능을 평가한 비무작위 배정, 공개 표지 시험이었습니다. 이러한 환자 중 아자시티딘 병용 요법을 받은 67명과 데시타빈 병용 요법을 받은 13명은 75세 이상이거나 집중적인 유도 화학 요법을 사용할 수 없는 동반 질환이 있었습니다.
환자는 VENCLEXTA 400mg을 경구로 1일 1회, 증량 투여 일정을 완료한 후 [투여 및 관리(2.3)]를 참조하여 아자시티딘(75mg/m2를 1주기 1일 1일부터 시작하여 28일 주기의 1~7일에 정맥 주사 또는 피하 주사) 또는 데시타빈(20mg/m2를 1주기 1일 1일부터 시작하여 28일 주기의 1~5일에 정맥 주사)과 병용하여 투여받았습니다. 증량 단계에서 환자는 TLS 예방을 받았으며 모니터링을 위해 입원했습니다. 환자는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 치료를 계속했습니다. 골수 평가를 통해 1주기 치료 후 백혈병 폭발이 5% 미만이고 빈혈이 나타나는 경우, VENCLEXTA는 최대 14일 또는 ANC ≥500/마이크로리터 및 혈소판 수 ≥50 × 103/마이크로리터가 될 때까지 중단되었습니다. 아자시티딘 용량 감소는 혈액학적 독성 관리를 위해 임상 시험에서 시행되었습니다. [투여 및 관리(2.5)]를 참조하십시오. 데시타빈의 용량 감소는 임상 시험에서 시행되지 않았습니다. 기준선 인구 통계 및 질병 특성은 표 26에 나와 있습니다.
| 특성 | VENCLEXTA + 아자시티딘 N = 67 |
VENCLEXTA + 데시타빈 N = 13 |
| 연령, 년; 중앙값(범위) | 76 (61-90) | 75 (68-86) |
| 인종; % | ||
| 백인 | 87 | 77 |
| 흑인 또는 아프리카계 미국인 | 4.5 | 0 |
| 아시아인 | 1.5 | 0 |
| 하와이 원주민 또는 태평양 섬 주민 | 1.5 | 15 |
| 아메리카 원주민/알래스카 원주민 | 0 | 7.7 |
| 보고되지 않은 기타 | 6 | 0 |
| 남성; % | 60 | 38 |
| ECOG 수행 상태; % 0-1 2 3 |
64 33 3 |
92 7.7 0 |
| 질병 병력; % 신규 AML 이차 AML |
73 27 |
85 15 |
| 돌연변이 분석 검출a; % | ||
| TP53 | 15 | 31 |
| IDH1 또는 IDH2 | 27 | 0 |
| FLT3 | 16 | 23 |
| NPM1 | 19 | 15 |
| 세포유전학적 위험 감지b,c; % | ||
| 중간 | 64 | 38 |
| 불량 | 34 | 62 |
| 기준선 동반 질환d; % | ||
| 심각한 심장 질환 | 4.5 | 7.7 |
| 심각한 폐 질환 | 1.5 | 0 |
| 중등도 간 기능 저하 | 9 | 0 |
| 크레아티닌 청소율 <45 mL/min | 13 | 7.7 |
| ECOG = 동부 협력 종양학 그룹. a아자시티딘 그룹에서 분석에 충분한 샘플이 없는 환자 6명과 데시타빈 그룹에서 4명 포함. b2014년판 국립 종합 암 네트워크(NCCN) 위험 분류에 따름. c아자시티딘 그룹의 환자 1명에서 유사 분열 없음(형광 현미경 하이브리드화[FISH] 분석에 의해 유리한 위험 제외). d환자는 하나 이상의 동반 질환을 가질 수 있음. |
||
효능 결과는 표 27에 나와 있습니다.
| 효능 결과 | VENCLEXTA + 아자시티딘 N = 67 |
VENCLEXTA + 데시타빈 N = 13 |
| CR, n (%) | 29 (43) | 7 (54) |
| (95% CI) | (31, 56) | (25, 81) |
| CRh, n (%) | 12 (18) | 1 (7.7) |
| (95% CI) | (9.6, 29) | (0.2, 36) |
| CI = 신뢰 구간; CR = 완전 관해; CRh = 부분 혈액학적 회복을 동반한 완전 관해. | ||
VENCLEXTA와 아자시티딘 병용 투여군의 중간 추적 관찰 기간은 15.9개월(범위: 0.4~40.3개월)이었습니다. CR의 중간 지속 기간은 23.8개월(95% CI: 15.4, -)이었고, CR+CRh의 중간 지속 기간은 26.5개월(95% CI: 17.4, -)이었습니다.
VENCLEXTA와 데시타빈 병용 투여군의 중간 추적 관찰 기간은 11.0개월(범위: 0.7~38.8개월)이었습니다. CR의 중간 지속 기간은 12.7개월(95% CI: 1.4, -)이었고, CR+CRh의 중간 지속 기간은 12.7개월(95% CI: 1.4, 20.0)이었습니다.
CR 지속 기간은 CR이 처음으로 기록된 날짜부터 재발, 임상적 질병 진행 또는 질병 진행으로 인한 사망이 가장 먼저 발생한 날짜까지의 기간으로 정의됩니다. CR+CRh 지속 기간은 CR 또는 CRh가 처음으로 기록된 날짜부터 재발, 임상적 질병 진행 또는 질병 진행으로 인한 사망이 가장 먼저 발생한 날짜까지의 기간으로 정의됩니다.
VENCLEXTA와 아자시티딘 병용 투여를 받은 환자의 첫 CR 또는 CRh까지의 중간 시간은 1.0개월(범위: 0.7~8.9개월)이었습니다.
VENCLEXTA와 데시타빈 병용 투여를 받은 환자의 첫 CR 또는 CRh까지의 중간 시간은 1.9개월(범위: 0.8~4.2개월)이었습니다.
VENCLEXTA와 아자시티딘 병용 투여를 받은 환자 중 12%(8/67)는 이후에 줄기세포 이식을 받았습니다.
이 임상시험에는 집중적인 유도 화학 요법을 사용할 수 없는 것으로 알려진 동반 질환이 없는 추가 환자 35명(연령 범위: 65~74세)이 등록되었으며, 이들은 VENCLEXTA와 아자시티딘(N=17) 또는 데시타빈(N=18) 병용 투여를 받았습니다.
VENCLEXTA와 아자시티딘 병용 투여를 받은 17명의 환자에서 CR율은 35%(95% CI: 14%, 62%)였습니다. CRh율은 41%(95% CI: 18%, 67%)였습니다. 9명(53%)의 환자가 이후에 줄기세포 이식을 받았습니다.
VENCLEXTA와 데시타빈 병용 투여를 받은 18명의 환자에서 CR율은 56%(95% CI: 31%, 79%)였습니다. CRh율은 22%(95% CI: 6.4%, 48%)였습니다. 4명(22%)의 환자가 이후에 줄기세포 이식을 받았습니다.
저용량 시타라빈과 병용 투여
VIALE-C는 저용량 시타라빈(VEN+LDAC)과 병용 투여한 VENCLEXTA의 효능과 안전성을 저용량 시타라빈(PBO+LDAC)과 병용 투여한 위약과 비교 평가한 무작위 배정(2:1), 이중 맹검, 위약 대조, 다기관 임상시험(NCT03069352)이었습니다.
환자는 증량 투여 일정을 완료한 후 1일 1회 경구로 VENCLEXTA 600mg을 1~28일에 투여받았습니다. [용법 및 용량(2.3)] 또는 위약을 1일 1회 피하로 시타라빈 20mg/m2와 병용 투여했습니다. 1일 1회, 28일 주기마다 1주기 1일부터 시작했습니다. 증량 단계에서 환자는 TLS 예방을 받았고, 모니터링을 위해 입원했습니다.
골수 평가를 통해 1주기 치료 후 5% 미만의 백혈병 폭발과 혈구 감소증이 확인되면, 즉, 완화가 확인되면, VENCLEXTA 또는 위약을 최대 14일까지 중단하거나, ANC ≥500/마이크로리터 및 혈소판 수 ≥50 × 103/마이크로리터가 될 때까지 중단했습니다. 1주기 말에 내성 질환이 있는 환자의 경우, 2주기 또는 3주기 후에 골수 평가를 실시하고 임상적으로 필요에 따라 실시했습니다. VENCLEXTA 또는 위약 중단 후 같은 날에 LDAC를 재개했습니다. 환자는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 치료를 계속했습니다. 기준 인구 통계적 특징과 질병 특징은 표 28에 나와 있습니다.
| 특징 | VENCLEXTA + 저용량 시타라빈 N = 143 |
위약 + 저용량 시타라빈 N = 68 |
| 연령, 년; 중간값(범위) | 76 (36, 93) | 76 (41, 88) |
| 인종; % | ||
| 백인 | 71 | 69 |
| 흑인 또는 아프리카계 미국인 | 1.4 | 1.5 |
| 아시아인 | 27 | 29 |
| 남성; % | 55 | 57 |
| ECOG 수행 상태; % 0-1 2 3 |
52 44 4.2 |
50 37 13 |
| 질병 병력; % 신규 AML 이차성 AML |
59 41 |
66 34 |
| 돌연변이 분석 검출; n/Na (%) | ||
| TP53 | 22/112 (20) | 9/52 (17) |
| IDH1 또는 IDH2 | 21/112 (19) | 12/52 (23) |
| FLT3 | 20/112 (18) | 9/52 (17) |
| NPM1 | 18/112 (16) | 7/52 (13) |
| Cytogenetic risk detectedb; % | ||
| Favorable | <1 | 4 |
| Intermediate | 63 | 63 |
| Poor | 33 | 29 |
| aNumber of evaluable BMA specimens received at baseline. bPer the 2016 National Comprehensive Cancer Network (NCCN) Guidelines. |
||
효능은 CR 비율 및 CR 지속 기간을 기반으로 하였으며, CR+CRh 비율, CR+CRh 지속 기간 및 수혈 의존에서 수혈 독립으로의 전환 비율에 대한 뒷받침 증거를 포함합니다. VEN+LDAC 군의 CR 비율은 27%(95% CI: 20%, 35%)였으며, CR의 중간 지속 기간은 11.1개월(95% CI: 6.1, -)이었고, PBO+LDAC 군의 CR 비율은 7.4%(95% CI: 2.4%, 16%)였으며, CR의 중간 지속 기간은 8.3개월(95% CI: 3.1, -)이었습니다. VEN+LDAC 군의 CR+CRh 비율은 47%(95% CI: 39%, 55%)였고, PBO+LDAC 군의 CR+CRh 비율은 15%(95% CI: 7.3%, 25%)였으며, VEN+LDAC 치료를 받은 경우 CR+CRh의 중간 지속 기간은 11.1개월이었고, PBO+LDAC 치료를 받은 경우 6.2개월이었습니다. CR 또는 CRh에 대한 첫 번째 반응까지의 중간 시간은 VEN+LDAC 치료를 받은 경우 1.0개월(범위: 0.7~5.8개월)이었습니다.
VEN+LDAC로 치료받은 환자 중 111명은 기준선에서 RBC 및/또는 혈소판 수혈에 의존했습니다. 이러한 환자 중 33%(37/111)명은 기준선 이후 연속적인 ≥56일 기간 동안 RBC 및 혈소판 수혈에서 독립되었습니다. VEN+LDAC로 치료받은 환자 중 32명은 기준선에서 RBC 및 혈소판 수혈 모두에서 독립적이었으며, 50%(16/32)명은 기준선 이후 연속적인 ≥56일 기간 동안 수혈 독립을 유지했습니다.
PBO+LDAC로 치료받은 환자 중 55명은 기준선에서 RBC 및/또는 혈소판 수혈에 의존했습니다. 이러한 환자 중 13%(7/55)명은 기준선 이후 연속적인 ≥56일 기간 동안 RBC 및 혈소판 수혈에서 독립되었습니다. PBO+LDAC로 치료받은 환자 중 13명은 기준선에서 RBC 및 혈소판 수혈 모두에서 독립적이었으며, 31%(4/13)명은 기준선 이후 연속적인 ≥56일 기간 동안 수혈 독립을 유지했습니다.
VEN+LDAC는 PBO+LDAC에 비해 OS를 유의하게 개선하지 못했습니다. OS에 대한 위험 비율(HR)은 0.75(95% CI: 0.52, 1.07)였으며, p-값은 0.114였습니다. VEN+LDAC 군의 중간 OS는 7.2개월(95% CI: 5.6, 10.1)이었고, PBO+LDAC 군의 중간 OS는 4.1개월(95% CI: 3.1, 8.8)이었습니다.
M14-387
M14-387(NCT02287233)은 이전에 선행 혈액 질환에 대한 저메틸화제에 노출된 환자를 포함하여 새로 진단된 AML 환자에서 VEN+LDAC(N=82)의 효능을 평가한 비무작위 배정, 공개 표지 시험이었습니다. 이러한 환자 중 61명은 75세 이상이었거나 집중적 유도 화학 요법을 사용할 수 없는 동반 질환이 있었습니다.
환자는 증량 단계가 완료된 후 1일 1회 경구로 VENCLEXTA 600mg을 1~28일에 투여받았습니다. [용법 및 용량(2.3)] 사이클 1일 1일에 시작하여 각 28일 사이클의 1~10일에 1일 1회 피하로 시타라빈 20mg/m2를 병용 투여했습니다. 증량 기간 동안 환자는 TLS 예방을 받았으며 모니터링을 위해 입원했습니다. 골수 평가를 통해 사이클 1 치료 후 5% 미만의 백혈병 폭발과 빈혈이 확인되면, 즉 완화가 확인되면 VENCLEXTA는 최대 14일까지 또는 ANC ≥500/마이크로리터 및 혈소판 수 ≥50 × 103/마이크로리터가 될 때까지 중단되었습니다. 환자는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 치료를 계속했습니다. 기준선 인구 통계 및 질병 특징은 표 29에 나와 있습니다.
| 특징 | 저용량 시타라빈과 병용한 VENCLEXTA N = 61 |
|
| 연령, 년; 중간값(범위) | 76 (63-90) | |
| 인종; % | ||
| 백인 | 92 | |
| 흑인 또는 아프리카계 미국인 | 1.6 | |
| 아시아인 | 1.6 | |
| 보고되지 않음 | 4.9 | |
| 남성; % | 74 | |
| ECOG 수행 상태; % 0-1 2 3 |
66 33 1.6 |
|
| 질병 병력; % 새로운 AML 이차 AML |
54 46 |
|
| 돌연변이 분석 검출a; % | ||
| TP53 | 8.2 | |
| IDH1 또는 IDH2 | 23 | |
| FLT3 | 21 | |
| NPM1 | 9.8 | |
| 세포 유전학적 위험 검출b; % | ||
| 중간 | 59 | |
| 불량 | 34 | |
| 무분열 | 6.6 | |
| 기준선 동반 질환c; % | ||
| 심각한 심장 질환 | 9.8 | |
| 중등도 간 기능 장애 | 4.9 | |
| 크레아티닌 청소율 ≥30 또는 <45 mL/min | 3.3 |
| a분석에 필요한 샘플이 부족한 환자 7명 포함. bNational Comprehensive Cancer Network (NCCN) 위험 분류 v2014에 따름. c환자는 하나 이상의 동반 질환이 있을 수 있음. |
|
중간 추적 관찰 기간은 7.3개월(범위: 0.3~54.0개월)이었습니다. CR율은 21%(95% CI: 12, 34)였고 CRh율은 21%(95% CI: 12, 34)였습니다.
CR의 중간 지속 기간은 22.9개월(95% CI: 5.1, -)이었고 CR+CRh의 중간 지속 기간은 14.3개월(95% CI: 6.1, 31.2)이었습니다.
VEN+LDAC로 치료받은 환자의 첫 CR 또는 CRh까지의 중간 시간은 1.0개월(범위: 0.8~9.4개월)이었습니다.
이 시험에는 집중적인 유도 화학 요법을 사용할 수 없게 하는 알려진 동반 질환이 없는 추가 환자 21명(연령 범위: 67~74세)이 등록되었으며 VEN+LDAC로 치료받았습니다. CR율은 33%(95% CI: 15%, 57%)였습니다. CRh율은 24%(95% CI: 8.2%, 47%)였습니다. 한 명의 환자(4.8%)가 이후에 줄기 세포 이식을 받았습니다.
16 제공/보관 및 취급 방법
VENCLEXTA는 다음과 같이 제공됩니다.
| 포장 형태 | 정제 수 | 국가 의약품 코드 (NDC) |
| CLL/SLL 시작 패키지 | 각 패키지에는 4개의 주간 지갑형 블리스터 패키지가 포함되어 있습니다.
|
0074-0579-28 |
| 10mg 정제가 들어 있는 지갑 | 10mg 정제 14개 | 0074-0561-14 |
| 50mg 정제가 들어 있는 지갑 | 50mg 정제 7개 | 0074-0566-07 |
| 10mg 정제가 들어 있는 단일 용량 블리스터 | 10mg 정제 2개 | 0074-0561-11 |
| 50mg 정제가 들어 있는 단일 용량 블리스터 | 50mg 정제 1개 | 0074-0566-11 |
| 100mg 정제가 들어 있는 단일 용량 블리스터 | 100mg 정제 1개 | 0074-0576-11 |
| 100mg 정제가 들어 있는 병 | 100mg 정제 28개 | 0074-0576-30 |
| 100mg 정제가 들어 있는 병 | 100mg 정제 120개 | 0074-0576-22 |
VENCLEXTA 10mg 필름 코팅 정제는 둥글고 양쪽으로 볼록하며 옅은 노란색으로 한쪽에는 “V”가, 다른 쪽에는 “10”이 각인되어 있습니다.
VENCLEXTA 50mg 필름 코팅 정제는 타원형이고 양쪽으로 볼록하며 베이지색으로 한쪽에는 “V”가, 다른 쪽에는 “50”이 각인되어 있습니다.
VENCLEXTA 100mg 필름 코팅 정제는 타원형이고 양쪽으로 볼록하며 옅은 노란색으로 한쪽에는 “V”가, 다른 쪽에는 “100”이 각인되어 있습니다.
원래 용기에 넣어 86°F(30°C) 이하에서 보관하십시오. 습기로부터 보호하기 위해 원래 용기에 넣어 환자에게 제공하십시오.
17 환자 상담 정보
환자에게 FDA 승인 환자 라벨(약물 안내)을 읽도록 알려주십시오.
종양 용해 증후군
환자에게 특히 치료 시작 시, 증량 단계에서, 그리고 중단 후 재개 시 TLS의 잠재적 위험을 알려주고, 이러한 사건과 관련된 증상(발열, 오한, 메스꺼움, 구토, 혼돈, 호흡 곤란, 발작, 불규칙적인 심장 박동, 어둡거나 흐린 소변, 비정상적인 피로, 근육통 및/또는 관절 불편함)이 나타나면 즉시 건강 관리 제공자(HCP)에게 평가를 위해 보고하도록 알려주십시오 [경고 및 주의 사항 (5.1)].
환자에게 TLS 위험을 줄이기 위해 VENCLEXTA를 복용하는 동안 매일 적절히 수분을 섭취하도록 알려주십시오. 권장되는 양은 하루 6~8잔(총 약 56온스)의 물입니다. 환자는 첫 번째 복용량 2일 전과 첫 번째 복용량 당일에, 그리고 복용량이 증가할 때마다 물을 마셔야 합니다 [용량 및 투여 (2.4)].
환자에게 혈액 검사 또는 기타 실험실 검사를 위해 예약된 약속을 지키는 것이 중요하다는 것을 알려주십시오 [용량 및 투여 (2.4)].
환자에게 TLS를 모니터링할 수 있도록 병원이나 의료 시설에서 VENCLEXTA를 복용해야 할 수도 있다는 것을 알려주십시오.
호중구 감소증
환자에게 발열이나 감염 증상이 나타나면 즉시 HCP에게 연락하도록 알려주십시오. 환자에게 혈액 수치를 주기적으로 모니터링해야 할 필요성을 알려주십시오 [경고 및 주의 사항 (5.2)].
감염
환자에게 발열이나 감염 증상이 나타나면 즉시 HCP에게 연락하도록 알려주십시오 [경고 및 주의 사항 (5.3)].
약물 상호 작용
환자에게 VENCLEXTA 치료 중에 자몽, 세비야 오렌지 또는 별 열매를 섭취하지 않도록 알려주십시오. 환자에게 VENCLEXTA가 일부 약물과 상호 작용할 수 있으므로, 처방약, 일반 의약품, 비타민 및 허브 제품 사용을 건강 관리 제공자에게 알리도록 알려주십시오 [금기 사항 (4) 및 약물 상호 작용 (7.1)].
예방 접종
환자에게 VENCLEXTA 치료 중에는 생백신으로 예방 접종을 하지 않도록 알려주십시오. 생백신은 안전하지 않거나 효과적이지 않을 수 있습니다 [경고 및 주의 사항 (5.4)].
태아 독성
임산부에게 태아에 대한 잠재적 위험을 알려주십시오. 임신 가능성이 있는 여성에게 알려진 임신 또는 의심되는 임신을 건강 관리 제공자에게 알리도록 알려주십시오 [경고 및 주의 사항 (5.5) 및 특정 인구 집단에서의 사용 (8.1)].
임신 가능성이 있는 여성에게 치료 기간 동안과 마지막 복용량 후 30일 동안 효과적인 피임법을 사용하도록 알려주십시오 [특정 인구 집단에서의 사용 (8.3)].
수유
여성에게 VENCLEXTA 치료 중과 마지막 복용량 후 1주일 동안 모유 수유를 하지 않도록 알려주십시오 [특정 인구 집단에서의 사용 (8.2)].
불임
임신 가능성이 있는 남성에게 VENCLEXTA가 생식 능력을 손상시킬 수 있다는 것을 알려주십시오 [특정 인구 집단에서의 사용 (8.3)].
투여
환자에게 VENCLEXTA를 원래 용기에 보관하도록 알려주십시오.
환자에게 VENCLEXTA를 처방대로 정확히 복용하고, HCP의 지시가 없는 한 복용량을 변경하거나 VENCLEXTA 복용을 중단하지 않도록 알려주십시오. 환자에게 HCP의 지시에 따라 매일 약 같은 시간에 VENCLEXTA를 경구로 1회 복용하고, 정제를 씹거나, 부수거나, 쪼개지 않고 식사와 함께 물과 함께 삼켜야 한다는 것을 알려주십시오 [용량 및 투여 (2.8)].
CLL/SLL 환자에게 치료 첫 4주 동안 VENCLEXTA를 원래 포장에 보관하고, 정제를 다른 용기에 옮기지 않도록 알려주십시오.
환자에게 VENCLEXTA 복용량을 놓친 지 8시간 미만이면 즉시 놓친 복용량을 복용하고 다음 복용량은 평소대로 복용하도록 알려주십시오. VENCLEXTA 복용량을 놓친 지 8시간 이상이면 환자에게 기다렸다가 다음 복용량을 평소 시간에 복용하도록 알려주십시오 [용량 및 투여 (2.8)].
환자에게 VENCLEXTA 복용 후 구토를 하면 그날 추가 복용량을 복용하지 말고, 다음 날 평소 시간에 다음 복용량을 복용하도록 알려주십시오.
제조 및 판매:
AbbVie Inc.
North Chicago, IL 60064
및
판매:
Genentech USA, Inc.
A Member of the Roche Group
South San Francisco, CA 94080-4990
© 2024 AbbVie. 모든 권리 보유.
Venclexta 및 해당 디자인은 AbbVie Inc.의 상표입니다.
© 2024 Genentech, Inc. 모든 권리 보유.
20087124 R1
약물 안내문
| 투약 안내 VENCLEXTA® (벤클렉스타) (베네토클락스 정제) |
|
| VENCLEXTA에 대해 알아야 할 가장 중요한 정보는 무엇입니까? VENCLEXTA는 다음을 포함한 심각한 부작용을 일으킬 수 있습니다.
|
|
| ○ 발열 ○ 오한 ○ 메스꺼움 ○ 구토 ○ 혼란 ○ 호흡 곤란 |
○ 발작 ○ 불규칙적인 심장 박동 ○ 어둡거나 흐린 소변 ○ 비정상적인 피로 ○ 근육이나 관절 통증 |
| VENCLEXTA 치료 중에 TLS 위험을 줄이기 위해 충분한 물을 마시십시오. 첫 번째 복용 2일 전부터, 첫 번째 VENCLEXTA 복용 당일, 그리고 복용량이 증가할 때마다 매일 6~8잔 (총 약 56온스)의 물을 마시십시오. 부작용이 발생하면 의료진이 VENCLEXTA 복용을 연기하거나, 복용량을 줄이거나, 치료를 중단할 수 있습니다. 1주일 이상 중단 후 VENCLEXTA를 다시 시작할 때, 의료진은 TLS 위험을 다시 확인하고 복용량을 변경할 수 있습니다. 부작용에 대한 자세한 내용은 “VENCLEXTA의 가능한 부작용은 무엇입니까?“를 참조하십시오. |
|
| VENCLEXTA는 무엇입니까? VENCLEXTA는 다음과 같은 경우에 사용되는 처방약입니다.
VENCLEXTA가 어린이에게 안전하고 효과적인지 여부는 알려져 있지 않습니다. |
|
| 누가 VENCLEXTA를 복용해서는 안 됩니까? 종양 용해 증후군 (TLS) 위험 증가로 인해 VENCLEXTA를 처음 복용할 때와 복용량이 천천히 증가하는 동안에는 특정 약을 복용해서는 안 됩니다.
|
|
VENCLEXTA를 복용하기 전에 다음과 같은 경우를 포함하여 모든 의학적 상태에 대해 의료진에게 알리십시오.
복용하는 모든 약물에 대해 의료진에게 알리십시오. 여기에는 처방약과 일반 의약품, 비타민 및 허브 보충제가 포함됩니다. VENCLEXTA와 다른 약물은 서로 영향을 미쳐 심각한 부작용을 일으킬 수 있습니다. “누가 VENCLEXTA를 복용해서는 안 됩니까?”를 참조하십시오. |
|
VENCLEXTA는 어떻게 복용해야 합니까?
|
|
| VENCLEXTA를 복용하는 동안 무엇을 피해야 합니까? VENCLEXTA를 복용하는 동안 자몽 주스, 자몽, 세비야 오렌지(종종 마멀레이드에 사용됨) 또는 별 열매를 먹거나 마시지 마십시오. 이러한 제품은 혈액 내 VENCLEXTA의 양을 증가시킬 수 있습니다. |
|
| VENCLEXTA의 가능한 부작용은 무엇입니까? VENCLEXTA는 다음을 포함한 심각한 부작용을 유발할 수 있습니다.
VENCLEXTA 치료 중에 발열이나 감염 징후가 있는 경우 즉시 의료 서비스 제공자에게 알리십시오. |
|
|
|
|
AML 환자에서 아자시티딘 또는 데시타빈 또는 저용량 시타라빈과 병용했을 때 VENCLEXTA의 가장 흔한 부작용은 다음과 같습니다. |
|
|
|
| VENCLEXTA는 남성의 생식 능력에 문제를 일으킬 수 있습니다. 이는 자녀를 낳는 능력에 영향을 미칠 수 있습니다. 생식 능력에 대한 우려 사항이 있는 경우 의료 서비스 제공자와 상담하십시오. 이것들은 VENCLEXTA의 모든 가능한 부작용이 아닙니다. 부작용에 대한 의학적 조언은 의사에게 문의하십시오. FDA에 부작용을 보고할 수 있습니다. 1-800-FDA-1088. |
|
VENCLEXTA는 어떻게 보관해야 합니까?
VENCLEXTA 및 모든 의약품을 어린이의 손이 닿지 않는 곳에 보관하십시오. |
|
| VENCLEXTA의 안전하고 효과적인 사용에 대한 일반 정보. 의약품은 때때로 약물 안내서에 나열된 목적 이외의 목적으로 처방됩니다. VENCLEXTA는 처방되지 않은 질환에 사용하지 마십시오. VENCLEXTA를 다른 사람에게 주지 마십시오. 같은 증상이 있어도 해로울 수 있습니다. 의료 서비스 제공자 또는 약사에게 의료 전문가를 위해 작성된 VENCLEXTA에 대한 정보를 문의할 수 있습니다. |
|
| VENCLEXTA의 성분은 무엇입니까? 활성 성분: venetoclax 비활성 성분: copovidone, colloidal silicon dioxide, polysorbate 80, sodium stearyl fumarate, and calcium phosphate dibasic. 10 mg 및 100 mg 코팅 정제에는 iron oxide yellow, polyvinyl alcohol, polyethylene glycol, talc 및 titanium dioxide가 포함됩니다. 50 mg 코팅 정제에는 iron oxide yellow, iron oxide red, iron oxide black, polyvinyl alcohol, talc, polyethylene glycol 및 titanium dioxide가 포함됩니다. |
|
| 제조 및 판매: AbbVie Inc. North Chicago, IL 60064 © 2024 AbbVie. 모든 권리 보유. Venclexta 및 해당 디자인은 AbbVie Inc.의 상표입니다. 20087124 R1 |
판매: Genentech USA, Inc. A Member of the Roche Group South San Francisco, CA 94080-4990 © 2024 Genentech, Inc. 모든 권리 보유. |
| 자세한 내용은 www.venclexta.com을 방문하거나 1-800-633-9110으로 전화하십시오. | |
| 이 약물 안내서는 미국 식품의약국에서 승인했습니다. | 개정: 7/2024 |
주요 디스플레이 패널
NDC 0074-0579-28
CLL/SLL 시동팩
VENCLEXTA®
(베네토클락스 정제)
10 mg, 50 mg, 및 100 mg
시동팩
! 경고
이 약을 받으시면 의사와 연락하십시오.
잠재적인 심각한 부작용을 예방하기 위해 의사의 입회 하에 첫 번째 복용량을 복용해야 할 수 있습니다.
조제사: VENCLEXTA를 조제할 때마다 환자에게 동봉된 약물 안내서를 제공하십시오.
abbvie
처방전에 의해서만
Genentech
주요 디스플레이 패널
NDC 0074-0561-14
Rx only
VENCLEXTA®
(venetoclax tablets)
10 mg per tablet
14 Tablets
Dispense the accompanying Medication Guide to each patient.
abbvie
Genentech

주요 디스플레이 패널
NDC 0074-0566-11
Rx only
VENCLEXTA®
(venetoclax tablets)
50 mg 정당
1정
각 환자에게 동봉된 복약 안내서를 제공하십시오.
abbvie
Genentech
주요 디스플레이 패널
NDC 0074-0576-22
처방전에 의해서만
VENCLEXTA®
(베네토클렉스 정제)
100 mg
주의: Venclexta는 원래 용기에 보관하여 습기로부터 보호하십시오.
120 정
동봉된 약물 안내서를 각 환자에게 제공하십시오.
abbvie
Genetech
