
의약품 제조업체: Gilead Sciences (Updated: 2024-12-02)
처방 정보 하이라이트
정맥 투여용 TRODELVY® (사시투주맙 고비테칸-hziy) 주사제
미국 최초 승인: 2020
경고: 호중구 감소증 및 설사
전체 경고 상자에 대한 자세한 내용은 전체 처방 정보를 참조하십시오.
중증 또는 생명을 위협하는 호중구 감소증이 발생할 수 있습니다. 절대 호중구 수가 1500/mm3 미만이거나 호중구 감소증 발열이 있는 경우 TRODELVY 투여를 중단하십시오. 치료 중에 정기적으로 혈구 수를 모니터링하십시오. 이차 예방을 위해 G-CSF를 고려하십시오. 발열성 호중구 감소증 환자에게는 지체 없이 항감염 치료를 시작하십시오. (2.3, 5.1)
중증 설사가 발생할 수 있습니다. 설사 환자를 모니터링하고 필요에 따라 수분과 전해질을 공급하십시오. 설사 발생 시 감염 원인을 평가하고, 감염이 없는 경우 즉시 로페라미드를 시작하십시오. 중증 설사가 발생하는 경우, 1등급 이하로 회복될 때까지 TRODELVY 투여를 중단하고 그 후 용량을 줄이십시오. (2.3, 5.2)
최근 주요 변경 사항
| 적응증 및 용법, 국소 진행성 또는 전이성 요로상피암 – 가속 승인 (텍스트 삭제) (1.2) | 2024년 11월 |
적응증 및 용법
TRODELVY는 Trop-2 표적 항체 및 토포이소머라제 억제제 접합체로 다음 환자의 치료에 사용됩니다.
국소 진행성 또는 전이성 유방암
용법 및 용량
- 이리노테칸 또는 그 활성 대사체인 SN-38을 함유한 다른 약물과 TRODELVY를 대체하거나 함께 사용하지 마십시오. (2.1)
- 정맥 주입 전용입니다. 정맥 푸시 또는 볼러스로 투여하지 마십시오.
- 권장 용량은 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 21일 연속 치료 주기의 1일차와 8일차에 1주일에 한 번 10mg/kg입니다. (2.2)
- 주입 반응 예방 및 항암제 유발 오심 및 구토 예방을 위한 전처치가 권장됩니다. (2.2)
- 주입 중 및 주입 완료 후 최소 30분 동안 환자를 모니터링하십시오. 유해 반응을 관리하기 위해 치료 중단 및/또는 용량 감소가 필요할 수 있습니다. (2.2)
- 제조 및 투여 지침에 대한 전체 처방 정보를 참조하십시오. (2.4)
제형 및 강도
주사제: 재구성을 위한 1회용 바이알에 동결 건조된 180mg 분말. (3)
경고 및 주의 사항
- 과민 반응 및 주입 관련 반응: 중증 아나필락시스 반응을 포함한 과민 반응이 관찰되었습니다. 주입 관련 반응에 대해 환자를 모니터링하십시오. 중증 또는 생명을 위협하는 반응이 발생하는 경우 TRODELVY 투여를 영구적으로 중단하십시오. (5.3)
- 오심/구토: 항구토제 예방 치료를 사용하고 예정된 치료 시 3등급 오심 또는 3~4등급 구토가 있는 환자의 경우 TRODELVY 투여를 중단하십시오. (5.4)
- UGT1A1 활성이 감소된 환자: 유리딘 이인산-글루쿠로노실 전달효소 1A1 (UGT1A1)*28 대립 유전자에 대해 동형 접합인 개인은 TRODELVY 치료 시작 후 호중구 감소증, 발열성 호중구 감소증 및 빈혈 위험이 증가합니다. (5.5)
- 배아-태아 독성: TRODELVY는 태아에게 해를 끼칠 수 있습니다. 환자에게 태아에 대한 잠재적 위험과 효과적인 피임법 사용에 대해 알리십시오. (5.6, 8.1, 8.3)
유해 반응
가장 흔한 유해 반응 (발생률 ≥25%)은 (실험실 이상 포함) 백혈구 수 감소, 호중구 수 감소, 헤모글로빈 감소, 설사, 오심, 림프구 수 감소, 피로, 탈모, 변비, 혈당 증가, 알부민 감소, 구토, 식욕 감소, 크레아티닌 청소율 감소, 알칼리성 인산가수분해효소 증가, 마그네슘 감소, 칼륨 감소 및 나트륨 감소였습니다. (6.1)
의심되는 유해 반응을 보고하려면 Gilead Sciences, Inc.에 1-888-983-4668 또는 FDA에 1-800-FDA-1088 또는 www.fda.gov/medwatch로 연락하십시오.
환자 상담 정보 및 FDA 승인 환자 라벨링은 17번을 참조하십시오.
개정: 2024년 11월
목차
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: 호중구감소증 및 설사
1 적응증 및 용법
1.1 국소 진행성 또는 전이성 유방암
2 용량 및 투여
2.1 중요 사용 정보
2.2 권장 용량
2.3 이상반응에 대한 용량 조정
2.4 조제 및 투여
3 제형 및 함량
4 금기
5 경고 및 주의사항
5.1 호중구감소증
5.2 설사
5.3 과민증 및 주입 관련 반응
5.4 메스꺼움 및 구토
5.5 UGT1A1 활성 감소 환자의 이상반응 위험 증가
5.6 배아-태아 독성
6 이상반응
6.1 임상시험 경험
7 약물 상호작용
7.1 다른 약물이 TRODELVY에 미치는 영향
8 특정 집단에서의 사용
8.1 임신
8.2 수유
8.3 가임기 여성 및 남성
8.4 소아에서의 사용
8.5 노인에서의 사용
8.6 간 장애
10 과다 복용
11 설명
12 임상 약리학
12.1 작용 기전
12.2 약력학
12.3 약동학
12.5 약물유전체학
12.6 면역원성
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 장애
14 임상 연구
14.1 국소 진행성 또는 전이성 삼중 음성 유방암
14.2 국소 진행성 또는 전이성 HR 양성, HER2 음성 유방암
15 참고문헌
16 공급/보관 및 취급 방법
17 환자 상담 정보
- *
- 전체 처방 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
제품에 대한 경고(BOXED WARNING)
경고: 호중구 감소증 및 설사
- 중증 또는 생명을 위협하는 호중구 감소증이 발생할 수 있습니다. 절대 호중구 수가 1500/mm3 미만이거나 호중구 감소증을 동반한 발열이 있는 경우 TRODELVY 투여를 중단하십시오. 치료 중에 정기적으로 혈구 수를 모니터링하십시오. 2차 예방을 위해 G-CSF를 고려하십시오 [see Dosage and Administration (2.3)]. 발열성 호중구 감소증 환자에게는 지체 없이 항감염 치료를 시작하십시오 [see Warnings and Precautions (5.1)].
- 중증 설사가 발생할 수 있습니다. 설사 환자를 모니터링하고 필요에 따라 수액 및 전해질을 투여하십시오. 설사 발생 시 감염 원인을 평가하고, 감염이 아닌 경우 즉시 로페라미드를 시작하십시오 [see Warnings and Precautions (5.2)]. 중증 설사가 발생하는 경우, 1등급 이하로 호전될 때까지 TRODELVY 투여를 중단하고 그 후의 용량을 감소시키십시오 [see Dosage and Administration (2.3)].
1 적응증 및 사용법
1.1 국소 진행성 또는 전이성 유방암
- TRODELVY는 절제 불가능한 국소 진행성 또는 전이성 삼중 음성 유방암(mTNBC) 성인 환자 중 전신 요법을 두 번 이상 받았으며, 그 중 적어도 한 번은 전이성 질환에 대해 받은 환자의 치료에 적응증이 됩니다.
- TRODELVY는 절제 불가능한 국소 진행성 또는 전이성 호르몬 수용체(HR) 양성, 사람 상피세포 성장인자 수용체 2(HER2) 음성(IHC 0, IHC 1+ 또는 IHC 2+/ISH–) 유방암 성인 환자 중 내분비 기반 요법과 전이성 질환에 대해 두 가지 이상의 추가 전신 요법을 받은 환자의 치료에 적응증이 됩니다.
2 용법 및 투여
2.2 권장 용량
TRODELVY의 권장 용량은 21일 치료 주기의 1일과 8일에 10mg/kg을 정맥 주입으로 투여하는 것입니다. 질병이 진행되거나 허용할 수 없는 독성이 나타날 때까지 치료를 계속하십시오. TRODELVY를 10mg/kg을 초과하는 용량으로 투여하지 마십시오.
TRODELVY는 정맥 주입으로만 투여하십시오. 정맥내 주사 또는 bolus로 투여하지 마십시오.
첫 번째 주입: 3시간 동안 주입합니다. 주입 중 및 초기 용량 투여 후 최소 30분 동안 주입 관련 반응의 징후 또는 증상이 있는지 환자를 관찰합니다. [see Warning and Precautions (5.3)].
후속 주입: 이전 주입을 잘 견뎌냈다면 1~2시간 동안 주입합니다. 주입 중 및 주입 후 최소 30분 동안 환자를 관찰합니다.
2.3 이상 반응에 대한 용량 조정
주입 관련 반응
환자에게 주입 관련 반응이 발생하면 TRODELVY의 주입 속도를 늦추거나 중단합니다. 생명을 위협하는 주입 관련 반응이 발생하는 경우 TRODELVY를 영구적으로 중단합니다. [see Warnings and Precautions (5.3)]
이상 반응에 대한 용량 조정
표 1에 설명된 대로 이상 반응을 관리하기 위해 TRODELVY를 보류하거나 중단합니다. 이상 반응으로 인해 용량을 줄인 후에는 TRODELVY 용량을 다시 늘리지 마십시오.
| 이상 반응 | 발생 | 용량 조정 |
|---|---|---|
| 중증 호중구 감소증 [see Warnings and Precautions (5.1)] | ||
| 4등급 호중구 감소증 ≥ 7일 또는 3~4등급 열성 호중구 감소증, 또는 예정된 치료 시점에 3~4등급 호중구 감소증으로 인해 1등급 이하로 회복될 때까지 투약이 2주 또는 3주 지연되는 경우 |
첫 번째 | 25% 용량 감소 및 과립구 콜로니 자극 인자(G-CSF) 투여. |
| 두 번째 | 50% 용량 감소 및 G-CSF 투여. | |
| 세 번째 | 치료 중단 및 G-CSF 투여. | |
| 예정된 치료 시점에 3~4등급 호중구 감소증으로 인해 1등급 이하로 회복될 때까지 투약이 3주 이상 지연되는 경우 | 첫 번째 | 치료 중단 및 G-CSF 투여. |
| 중증 비호중구성 독성 | ||
| 기간에 관계없이 4등급 비혈액학적 독성, 또는 항구토제 및 지사제로 조절되지 않는 치료로 인한 3~4등급 메스꺼움, 구토 또는 설사 [see Warnings and Precautions (5.2, 5.4)], 또는 최적의 의학적 관리에도 불구하고 48시간 이상 지속되는 기타 3~4등급 비혈액학적 독성, 또는 예정된 치료 시점에 1등급 이하로 회복될 때까지 투약이 2주 또는 3주 지연되는 3~4등급 비호중구성 혈액학적 또는 비혈액학적 독성 |
첫 번째 | 25% 용량 감소 |
| 두 번째 | 50% 용량 감소 | |
| 세 번째 | 치료 중단 | |
| 3~4등급 비호중구성 혈액학적 또는 비혈액학적 독성이 3주 이내에 1등급 이하로 회복되지 않는 경우 | 첫 번째 | 치료 중단 |
2.4 준비 및 투여
재구성
- TRODELVY는 위험한 약물입니다.
- 해당되는 특수 취급 및 폐기 절차를 따르십시오1.
- 각 치료 주기 시작 시(또는 이전 투여 이후 환자의 체중이 10% 이상 변경된 경우 더 자주) 환자의 체중을 기준으로 TRODELVY의 필요한 용량(mg)을 계산합니다 [see Dosage and Administration (2.2)].
- 필요한 수의 바이알을 실온으로 데웁니다.
- 멸균 주사기를 사용하여 180mg TRODELVY 바이알 각각에 20mL의 0.9% 염화나트륨 주사액, USP를 천천히 주입합니다. 각 바이알에는 준비 과정 및 재구성 후 액체 손실을 보완하기 위한 초과 충전량이 포함되어 있으며, 결과적으로 총 부피는 10mg/mL의 농도를 제공합니다.
- 바이알을 부드럽게 흔들어 최대 15분 동안 용해시킵니다. 흔들지 마십시오. 비경구 약물은 용액과 용기가 허용하는 경우 투여 전에 미립자 물질과 변색 여부를 육안으로 검사해야 합니다. 용액은 눈에 보이는 입자가 없고 투명하고 노란색이어야 합니다. 재구성된 용액이 흐리거나 변색된 경우 사용하지 마십시오.
- 희석된 TRODELVY 주입 용액을 준비하려면 즉시 사용하십시오.
희석
- 환자의 체중에 따라 적절한 용량을 얻는 데 필요한 재구성된 TRODELVY 용액의 필요량을 계산합니다.
- 1.1mg/mL ~ 3.4mg/mL의 TRODELVY 농도 범위에서 적절한 용량을 전달하기 위한 주입 용액의 최종 부피를 결정합니다.
- 재구성된 TRODELVY 용액의 안정성은 다른 주입 기반 용액으로 결정되지 않았으므로 0.9% 염화나트륨 주사액, USP만 사용하십시오. 폴리염화비닐, 폴리프로필렌/폴리에틸렌, 폴리올레핀 또는 에틸렌 비닐 아세테이트 주입 백을 사용하십시오.
- 계산된 양의 재구성된 TRODELVY 용액을 첨가한 후 표시된 TRODELVY 농도를 달성하는 데 필요한 최종 주입 백에서 0.9% 염화나트륨 주사액, USP의 부피를 빼내고 버립니다.
- 주사기를 사용하여 바이알에서 계산된 양의 재구성된 TRODELVY 용액을 빼냅니다. 바이알에 남아 있는 사용하지 않은 부분은 버립니다.
- 거품 발생을 최소화하려면 계산된 양의 재구성된 TRODELVY 용액을 주입 백에 천천히 주입합니다. 내용물을 흔들지 마십시오.
- 즉시 사용하지 않는 경우, TRODELVY 용액이 들어 있는 주입 백은 빛으로부터 보호하여 2°C ~ 8°C(36°F ~ 46°F)에서 최대 24시간 동안 냉장 보관할 수 있습니다. 냉장 후 희석된 용액을 실온에서 최대 25°C(77°F)까지 8시간 이내(주입 시간 포함)에 투여하십시오.
냉동하거나 흔들지 마십시오.
3 제형 및 함량
주사용: 1회용 바이알에 180mg의 백색에서 황색을 띠는 동결건조 분말.
4 금기 사항
TRODELVY는 TRODELVY에 대한 중증 과민반응을 경험한 환자에게는 금기입니다 [경고 및 주의사항 (5.3) 참조].
5 경고 및 주의사항
5.1 호중구 감소증
TRODELVY로 치료받는 환자에게서 중증, 생명을 위협하거나 치명적인 호중구 감소증이 발생할 수 있습니다. TRODELVY로 치료받은 환자의 64%에서 호중구 감소증이 발생했습니다. 3-4등급 호중구 감소증은 환자의 49%에서 발생했습니다. 발열성 호중구 감소증은 환자의 6%에서 발생했습니다. 호중구 감소증(발열성 호중구 감소증 포함)의 최초 발병까지의 중앙값 시간은 16일이었으며 일부 환자군에서는 더 일찍 발생했습니다 [경고 및 주의사항 (5.5) 참조]. 호중구 감소성 대장염은 환자의 1.4%에서 발생했습니다.
어떤 주기의 1일차에 절대 호중구 수가 1500/mm3 미만이거나 어떤 주기의 8일차에 호중구 수가 1000/mm3 미만인 경우 TRODELVY 투여를 중단합니다. 호중구 감소증 발열의 경우 TRODELVY 투여를 중단합니다. 호중구 감소증으로 인해 용량 조절이 필요할 수 있습니다. 임상적으로 필요하거나 표 1에 명시된 대로 G-CSF를 투여합니다 [용법 및 용량 (2.3) 참조].
5.2 설사
TRODELVY는 중증 설사를 유발할 수 있습니다. TRODELVY로 치료받은 모든 환자의 64%에서 설사가 발생했습니다. TRODELVY로 치료받은 모든 환자의 11%에서 3-4등급 설사가 발생했습니다. 한 환자는 설사 후 장 천공이 발생했습니다. 탈수 및 그에 따른 급성 신손상으로 이어진 설사는 모든 환자의 0.7%에서 발생했습니다.
예정된 치료 투여 시점에 3-4등급 설사가 발생하면 TRODELVY 투여를 중단하고 1등급 이하로 회복되면 재개합니다 [용법 및 용량 (2.3) 참조].
설사 발생 시 감염 원인을 평가하고, 감염이 없는 경우 로페라미드를 4mg으로 초기 투여한 후 설사 발생 시마다 2mg씩 최대 1일 16mg까지 투여합니다. 설사가 해소된 후 12시간 후에 로페라미드 투여를 중단합니다. 임상적으로 필요한 경우 추가적인 지지 요법(예: 수액 및 전해질 대체)을 사용할 수도 있습니다.
TRODELVY 치료에 대한 과도한 콜린성 반응(예: 복통, 설사, 타액 분비 등)을 보이는 환자는 후속 치료를 위해 적절한 전처치(예: 아트로핀)를 받을 수 있습니다.
5.3 과민반응 및 주입 관련 반응
생명을 위협하는 아나필락시스 반응을 포함한 중증 과민반응이 TRODELVY 치료와 함께 발생했습니다. 심각한 징후 및 증상에는 심정지, 저혈압, 천명, 안면부종, 부종, 폐렴 및 피부 반응이 포함됩니다 [금기사항 (4) 참조].
투약 후 24시간 이내에 발생한 과민반응은 TRODELVY로 치료받은 환자의 35%에서 발생했습니다. 3-4등급 과민반응은 TRODELVY로 치료받은 환자의 2%에서 발생했습니다. TRODELVY의 영구적인 중단으로 이어진 과민반응의 발생률은 0.2%였습니다. 아나필락시스 반응의 발생률은 0.2%였습니다.
TRODELVY를 투여받는 환자의 주입 반응에 대한 전처치가 권장됩니다. TRODELVY를 투여할 때 아나필락시스를 포함한 주입 관련 반응을 치료하기 위한 약물과 응급 장비를 즉시 사용할 수 있도록 준비해야 합니다 [용법 및 용량 (2.2) 참조].
각 TRODELVY 주입 중 및 각 주입 완료 후 최소 30분 동안 과민반응 및 주입 관련 반응에 대해 환자를 면밀히 모니터링합니다 [용법 및 용량 (2.3) 참조].
4등급 주입 관련 반응의 경우 TRODELVY 투여를 영구적으로 중단합니다 [용법 및 용량 (2.3) 참조].
5.4 오심 및 구토
TRODELVY는 구토 유발성입니다. TRODELVY로 치료받은 모든 환자의 64%에서 오심이 발생했습니다. 3-4등급 오심은 환자의 3%에서 발생했습니다.
TRODELVY로 치료받은 모든 환자의 35%에서 구토가 발생했습니다. 이러한 환자 중 2%에서 3-4등급 구토가 발생했습니다.
항암 화학요법 유발 오심 및 구토(CINV) 예방을 위해 2가 또는 3가 약물 병용 요법(예: 덱사메타손과 5-HT3 수용체 길항제 또는 NK1 수용체 길항제 및 기타 필요한 약물)으로 전처치합니다 [용법 및 용량 (2.2) 참조].
예정된 치료 투여 시점에 3등급 오심 또는 3-4등급 구토가 발생하면 TRODELVY 용량 투여를 중단하고 추가적인 지지 요법을 통해 1등급 이하로 회복되면 재개합니다 [용법 및 용량 (2.3) 참조].
임상적으로 필요한 경우 추가적인 항구토제 및 기타 지지 요법을 사용할 수도 있습니다. 모든 환자에게 오심 및 구토 예방 및 치료에 대한 명확한 지침과 함께 가정용 약물을 제공해야 합니다.
5.5 UGT1A1 활성이 감소된 환자에서의 유해 반응 위험 증가
우리딘 이인산-글루쿠로노실 전이효소 1A1 (UGT1A1)*28 대립유전자에 대해 동형접합인 환자는 TRODELVY로 치료받을 때 호중구 감소증, 발열성 호중구 감소증 및 빈혈의 위험이 증가하고 다른 유해 반응의 위험이 증가할 수 있습니다.
TRODELVY를 투여받고 UGT1A1 유전자형 결과를 가진 948명의 환자에서 호중구 감소증 및 빈혈의 발생률을 분석했습니다. UGT1A1 *28 대립유전자에 대해 동형접합인 환자(n=112)의 경우 3-4등급 호중구 감소증의 발생률은 58%였습니다. UGT1A1*28 대립유전자에 대해 이형접합인 환자(n=420)의 경우 3-4등급 호중구 감소증의 발생률은 49%였습니다. 야생형 대립유전자에 대해 동형접합인 환자(n=416)의 경우 3-4등급 호중구 감소증의 발생률은 43%였습니다 [임상 약리학 (12.5) 참조]. UGT1A1 *28 대립유전자에 대해 동형접합인 환자의 경우 3-4등급 빈혈의 발생률은 21%였습니다. UGT1A1*28 대립유전자에 대해 이형접합인 환자의 경우 3-4등급 빈혈의 발생률은 10%였습니다. 야생형 대립유전자에 대해 동형접합인 환자의 경우 3-4등급 빈혈의 발생률은 9%였습니다.
UGT1A1*28 대립유전자 동형접합 환자의 첫 번째 호중구감소증(발열성 호중구감소증 포함)까지의 중앙값 시간은 9일, 이형접합 환자는 15일, 야생형 대립유전자 동형접합 환자는 20일이었습니다. UGT1A1*28 대립유전자 동형접합 환자의 첫 번째 빈혈까지의 중앙값 시간은 21일, 이형접합 환자는 25일, 야생형 대립유전자 동형접합 환자는 28일이었습니다.
알려진 UGT1A1 활성 감소 환자의 경우, 이상 반응을 면밀히 모니터링하십시오. 급성 조기 발현 또는 비정상적으로 심각한 이상 반응(이는 UGT1A1 효소 활성 감소를 나타낼 수 있음)이 나타나는 환자의 경우, 관찰된 이상 반응의 발현, 지속 기간 및 중증도에 대한 임상적 평가에 따라 TRODELVY 투여를 중단하거나 영구적으로 중단하십시오 [투여 및 투약 방법 (2.3) 참조].
5.6 배아-태아 독성
작용 기전에 근거하여, TRODELVY는 임신 여성에게 투여될 경우 기형성 및/또는 배아-태아 치사를 유발할 수 있습니다. TRODELVY는 유전독성 성분인 SN-38을 함유하고 있으며, 빠르게 분열하는 세포를 표적으로 합니다 [임상 약리학 (12.1) 및 비임상 독성학 (13.1) 참조]. 임신 여성과 임신 가능성이 있는 여성에게 태아에 대한 잠재적 위험을 알리십시오. 임신 가능성이 있는 여성에게는 TRODELVY 치료 기간 동안과 마지막 투여 후 6개월 동안 효과적인 피임법을 사용하도록 조언하십시오. 임신 가능성이 있는 여성 파트너가 있는 남성 환자에게는 TRODELVY 치료 기간 동안과 마지막 투여 후 3개월 동안 효과적인 피임법을 사용하도록 조언하십시오 [특정 집단에서의 사용 (8.1), 8.3) 참조].
6 부작용 반응
다음의 유해 반응은 이 약의 설명서 다른 부분에서 더 자세히 설명하고 있습니다:
- 호중구 감소증 [경고 및 주의사항 (5.1) 참조]
- 설사 [경고 및 주의사항 (5.2) 참조]
- 과민반응 및 주입 관련 반응 [경고 및 주의사항 (5.3) 참조]
- 오심 및 구토 [경고 및 주의사항 (5.4) 참조]
6.1 임상시험 경험
임상시험은 매우 다양한 조건 하에서 수행되므로, 특정 약물의 임상시험에서 관찰된 유해 반응 발생률을 다른 약물의 임상시험에서 관찰된 발생률과 직접 비교할 수 없으며, 임상 현장에서 관찰된 발생률을 반영하지 않을 수도 있습니다.
경고 및 주의사항 섹션에 설명된 통합 안전성 모집단은 IMMU-132-01, ASCENT 및 TROPiCS-02에서 mTNBC 환자 366명과 호르몬 수용체 양성(HR+)/인간 표피 성장 인자 수용체 2 음성(HER2-) 유방암 환자 322명을 포함한 1063명의 환자에게 TRODELVY를 투여한 결과를 반영합니다. 그리고 다른 종양 유형의 환자 375명을 포함합니다. TRODELVY는 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 21일 치료 주기의 1일차와 8일차에 10 mg/kg의 용량으로 1주일에 한 번 정맥 주입으로 투여되었습니다. TRODELVY로 치료받은 1063명의 환자 중 치료 기간 중앙값은 4.1개월(범위: 0~63개월)이었습니다. 이 통합 안전성 모집단에서 가장 흔한(≥25%) 유해 반응(실험실 이상 포함)은 백혈구 수 감소(84%), 호중구 수 감소(75%), 헤모글로빈 감소(69%), 설사(64%), 오심(64%), 림프구 수 감소(63%), 피로(51%), 탈모(45%), 변비(37%), 혈당 증가(37%), 알부민 감소(35%), 구토(35%), 식욕 감소(30%), 크레아티닌 청소율 감소(28%), 알칼리성 인산가수분해효소 증가(28%), 마그네슘 감소(27%), 칼륨 감소(26%) 및 나트륨 감소(26%)였습니다.
국소 진행성 또는 전이성 삼중 음성 유방암
ASCENT 연구
택산 및 2회 이상의 항암화학요법을 이전에 받은 적이 있는 mTNBC 환자를 대상으로 한 무작위 배정, 활성 대조, 공개 표지 연구(ASCENT)에서 TRODELVY의 안전성을 평가했습니다. 환자는 TRODELVY(n=258) 또는 단독 요법 항암화학요법(n=224)을 받도록 무작위 배정(1:1)되었으며, 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 치료를 받았습니다 [임상 연구(14.1) 참조]. TRODELVY로 치료받은 환자의 치료 기간 중앙값은 4.4개월(범위: 0~23개월)이었습니다.
TRODELVY를 투여받은 환자의 27%에서 중대한 유해 반응이 발생했습니다. TRODELVY를 투여받은 환자의 1% 이상에서 발생한 중대한 유해 반응에는 호중구 감소증(7%), 설사(4%) 및 폐렴(3%)이 포함됩니다. TRODELVY를 투여받은 환자의 1.2%에서 치명적인 유해 반응이 발생했으며, 여기에는 호흡 부전(0.8%) 및 폐렴(0.4%)이 포함됩니다. 유해 반응으로 인해 5%의 환자에서 TRODELVY 투여가 영구적으로 중단되었습니다. TRODELVY를 투여받은 환자의 1% 이상에서 영구 중단으로 이어진 유해 반응은 폐렴(1%) 및 피로(1%)였습니다.
TRODELVY 치료 중단으로 이어진 유해 반응은 환자의 63%에서 발생했습니다. 치료 중단으로 이어진 가장 빈번한(≥5%) 유해 반응은 호중구 감소증(47%), 설사(5%), 호흡기 감염(5%) 및 백혈구 감소증(5%)이었습니다.
TRODELVY 용량 감소로 이어진 유해 반응은 환자의 22%에서 발생했습니다. 용량 감소로 이어진 가장 빈번한(>4%) 유해 반응은 호중구 감소증(11%) 및 설사(5%)였습니다.
TRODELVY를 투여받은 환자의 44%에서 과립구 콜로니 자극 인자(G-CSF)가 사용되었습니다.
표 2와 표 3은 ASCENT 연구에서의 유해 반응과 실험실 이상을 각각 요약한 것입니다.
| TRODELVY (n=258) | 단독 요법 항암화학요법 (n=224) | |||
|---|---|---|---|---|
| 유해 반응 | 모든 등급 % |
3~4등급 % |
모든 등급 % |
3~4등급 % |
| *단독 요법 항암화학요법은 다음 단일 약제 중 하나를 포함했습니다: 에리불린(n=139), 카페시타빈(n=33), 젬시타빈(n=38) 또는 비노렐빈(환자가 2등급 이상의 신경병증이 없는 경우, n=52). | ||||
| NCI CTCAE v.5.0에 따라 등급이 매겨졌습니다. | ||||
| 위장관 장애 | ||||
| 설사 | 59 | 11 | 17 | 1 |
| 오심 | 57 | 3 | 26 | 0.4 |
| 구토 | 33 | 2 | 16 | 1 |
| 변비 | 37 | 0.4 | 23 | 0 |
| 복통 | 30 | 3 | 12 | 1 |
| 구내염* | 17 | 2 | 13 | 1 |
| 일반 장애 및 투여 부위 상태 | ||||
| 피로† | 65 | 6 | 50 | 9 |
| 발열 | 15 | 0.4 | 14 | 2 |
| 감염 및 기생충 감염 | ||||
| 요로 감염 | 13 | 0.4 | 8 | 0.4 |
| 상기도 감염 | 12 | 0 | 3 | 0 |
| 대사 및 영양 장애 | ||||
| 식욕 감퇴 | 28 | 2 | 21 | 1 |
| 근골격계 및 결합 조직 장애 | ||||
| 요통 | 16 | 1 | 14 | 2 |
| 관절통 | 12 | 0.4 | 7 | 0 |
| 신경계 장애 | ||||
| 두통 | 18 | 0.8 | 13 | 0.4 |
| 현기증 | 10 | 0 | 7 | 0 |
| 정신 장애 | ||||
| 불면증 | 11 | 0 | 5 | 0 |
| 호흡기, 흉부 및 종격동 장애 | ||||
| 기침 | 24 | 0 | 18 | 0.4 |
| 피부 및 피하 조직 장애 | ||||
| 탈모 | 47 | 0 | 16 | 0 |
| 발진 | 12 | 0.4 | 5 | 0.4 |
| 가려움증 | 10 | 0 | 3 | 0 |
| 이상 실험실 검사 결과 | TRODELVY (n=258) |
단독 화학요법 (n=224) |
||
|---|---|---|---|---|
| 모든 등급 (%) |
3-4등급 (%) |
모든 등급 (%) |
3-4등급 (%) |
|
| 혈액학 | ||||
| 헤모글로빈 감소 | 94 | 9 | 57 | 6 |
| 림프구 수 감소 | 88 | 31 | 40 | 24 |
| 백혈구 수 감소 | 86 | 41 | 53 | 25 |
| 호중구 수 감소 | 78 | 49 | 48 | 36 |
| 혈소판 수 감소 | 23 | 1.2 | 25 | 2.7 |
| 화학 | ||||
| 혈당 증가 | 49 | 2.3 | 43 | 2.8 |
| 칼슘 감소 | 36 | 1.6 | 21 | 1.4 |
| 마그네슘 감소 | 33 | 0.4 | 20 | 0 |
| 칼륨 감소 | 33 | 4.3 | 28 | 0.9 |
| 알부민 증가 | 32 | 0.8 | 25 | 1.4 |
| 아스파르트산 아미노전이효소 증가 | 27 | 1.2 | 32 | 1.4 |
| 알라닌 아미노전이효소 증가 | 26 | 1.2 | 26 | 1.8 |
| 알칼리성 인산가수분해효소 증가 | 26 | 0 | 17 | 0.5 |
| 인산 감소 | 26 | 7.8 | 20 | 3.3 |
| 나트륨 감소 | 22 | 0.4 | 17 | 0.5 |
| 젖산 탈수소효소 증가 | 18 | 0 | 22 | 0 |
| 혈당 감소 | 10 | 0 | 3.2 | 0 |
IMMU-132-01 연구
TRODELVY의 안전성은 전이성 질환에 대해 2종 이상의 항암 치료를 받은 적이 있는 mTNBC 환자 108명을 포함하여 mTNBC 및 기타 악성 종양 환자를 대상으로 한 단일군, 공개 표지 연구(IMMU-132-01)에서 평가되었습니다 [임상 연구(14.1) 참조]. TRODELVY는 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 21일 치료 주기의 1일차와 8일차에 1주일에 한 번 정맥 주입으로 최대 10mg/kg의 용량으로 투여되었습니다. 이 108명의 환자에서 중앙값 치료 기간은 5.1개월(범위: 0~51개월)이었습니다.
환자의 31%에서 중대한 이상 반응이 발생했습니다. TRODELVY를 투여받은 환자의 1% 이상에서 발생한 중대한 이상 반응에는 발열성 호중구 감소증(6%), 구토(5%), 오심(3%), 호흡곤란(3%), 설사(4%), 빈혈(2%), 흉막 삼출, 호중구 감소증, 폐렴, 탈수(각각 2%)가 포함됩니다.
이상 반응으로 인해 TRODELVY 투여가 영구적으로 중단된 환자는 2%였습니다. 영구 중단으로 이어진 이상 반응은 아나필락시스, 식욕 부진/피로, 두통(각각 0.9%)이었습니다. 환자의 45%가 치료 중단으로 이어지는 이상 반응을 경험했습니다. 치료 중단으로 이어지는 가장 흔한 이상 반응은 호중구 감소증(33%)이었습니다. TRODELVY로 치료받은 환자의 33%에서 용량 감소로 이어지는 이상 반응이 발생했으며, 24%는 1회 용량 감소, 9%는 2회 용량 감소를 경험했습니다. 용량 감소로 이어지는 가장 흔한 이상 반응은 호중구 감소증/발열성 호중구 감소증이었습니다.
표 4와 5는 IMMU-132-01 연구에서 mTNBC 환자의 ≥10%에서 발생한 이상 반응과 실험실 이상을 요약한 것입니다.
| 이상 반응 | TRODELVY (n=108) |
|||
|---|---|---|---|---|
| 1~4등급 (%) |
3~4등급 (%) |
|||
| NCI CTCAE v. 4.0에 따라 등급이 매겨짐 | ||||
|
||||
| 모든 이상 반응 | 100 | 71 | ||
| 위장 장애 | 95 | 21 | ||
| 오심 | 69 | 6 | ||
| 설사 | 63 | 9 | ||
| 구토 | 49 | 6 | ||
| 변비 | 34 | 1 | ||
| 복통* | 26 | 1 | ||
| 점막염† | 14 | 1 | ||
| 일반 장애 및 투여 부위 상태 | 77 | 9 | ||
| 피로‡ | 57 | 8 | ||
| 부종§ | 19 | 0 | ||
| 발열 | 14 | 0 | ||
| 대사 및 영양 장애 | 68 | 22 | ||
| 식욕 감퇴 | 30 | 1 | ||
| 탈수 | 13 | 5 | ||
| 피부 및 피하 조직 장애 | 63 | 4 |
| 탈모 | 38 | 0 |
| 발진¶ | 31 | 3 |
| 가려움증 | 17 | 0 |
| 건조한 피부 | 15 | 0 |
| 신경계 장애 | 56 | 4 |
| 두통 | 23 | 1 |
| 어지러움 | 22 | 0 |
| 신경병증# | 24 | 0 |
| 미각 이상 | 11 | 0 |
| 감염 및 기생충 감염 | 55 | 12 |
| 요로 감염 | 21 | 3 |
| 호흡기 감염Þ | 26 | 3 |
| 근골격계 및 결합 조직 장애 | 54 | 1 |
| 요통 | 23 | 0 |
| 관절통 | 17 | 0 |
| 사지 통증 | 11 | 0 |
| 호흡기, 흉부 및 종격동 장애 | 54 | 5 |
| 기침ß | 22 | 0 |
| 호흡 곤란à | 21 | 3 |
| 정신 장애 | 26 | 1 |
| 불면증 | 13 | 0 |
| 이상 실험실 검사 결과 | TRODELVY (n=108) |
|
|---|---|---|
| 모든 등급 (%) |
3-4등급 (%) |
|
| 혈액학 | ||
| 헤모글로빈 감소 | 93 | 6 |
| 백혈구 감소 | 91 | 26 |
| 호중구 감소 | 82 | 32 |
| 활성화 부분 트롬보플라스틴 시간 증가 | 60 | 12 |
| 혈소판 감소 | 30 | 3 |
| 화학 | ||
| 알칼리성 포스파타제 증가 | 57 | 2 |
| 마그네슘 감소 | 51 | 3 |
| 칼슘 감소 | 49 | 3 |
| 아스파르트 아미노트랜스퍼라제 증가 | 45 | 3 |
| 알부민 감소 | 39 | 1 |
| 알라닌 아미노트랜스퍼라제 증가 | 35 | 2 |
| 글루코스 증가 | 31 | 2.8 |
| 인산염 감소 | 29 | 5 |
| 마그네슘 감소 | 27 | 1.9 |
| 인산염 감소 | 27 | 6.5 |
| 나트륨 감소 | 25 | 4.7 |
| 칼륨 감소 | 24 | 3.7 |
| 글루코스 감소 | 19 | 2 |
국소 진행성 또는 전이성 HR-양성, HER2-음성 유방암
TROPiCS-02 연구
TRODELVY의 안전성은 절제 불가능한 국소 진행성 또는 전이성 HR-양성, HER2-음성 유방암 환자를 대상으로 한 무작위 배정, 활성 대조, 공개 표지 연구(TROPiCS-02)에서 평가되었습니다. 이 환자들은 어떤 치료 환경에서든 다음 치료 후 질병이 진행된 경우였습니다. CDK 4/6 억제제, 내분비 요법 및 택산; 환자들은 전이 환경에서 최소 두 가지 이상의 이전 화학 요법(신보조 또는 보조 환경에서 하나일 수 있음, 진행이 12개월 이내에 발생한 경우)을 받았습니다. 환자들은 TRODELVY(n=268) 또는 단일 요법 화학 요법(n=249)을 받도록 무작위 배정(1:1)되었으며 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 치료를 받았습니다 [임상 연구(14.2) 참조]. TRODELVY로 치료받은 환자의 경우 중앙값 치료 기간은 4.1개월(범위: 0~63개월)이었습니다.
TRODELVY를 투여받은 환자의 28%에서 중대한 이상 반응이 발생했습니다. TRODELVY를 투여받은 환자의 1% 이상에서 발생한 중대한 이상 반응에는 설사(5%), 발열성 호중구 감소증(4%), 호중구 감소증(3%), 복통, 대장염, 호중구 감소증성 대장염, 폐렴 및 구토(각각 2%)가 포함되었습니다. TRODELVY를 투여받은 환자의 2%에서 부정맥, COVID-19, 신경계 장애, 폐색전증 및 패혈성 쇼크(각각 0.4%)를 포함한 치명적인 이상 반응이 발생했습니다. 이상 반응으로 인해 TRODELVY 투여가 영구적으로 중단된 환자는 6%였습니다. TRODELVY를 투여받은 환자에서 영구적인 투여 중단으로 이어진 가장 빈번한(≥0.5%) 이상 반응은 무력증, 전반적인 신체 건강 악화 및 호중구 감소증(각각 0.7%)이었습니다.
TRODELVY 투여 중단으로 이어진 이상 반응은 환자의 66%에서 발생했습니다. 치료 중단으로 이어진 가장 빈번한(≥5%) 이상 반응은 호중구 감소증(50%)이었습니다.
TRODELVY 용량 감소로 이어진 이상 반응은 환자의 33%에서 발생했습니다. 용량 감소로 이어진 가장 빈번한(>5%) 이상 반응은 호중구 감소증(16%)과 설사(8%)였습니다. G-CSF는 TRODELVY를 투여받은 환자의 54%에서 사용되었습니다.
표 6과 7은 TROPiCS-02 연구에서의 이상 반응과 실험실 이상을 요약한 것입니다.
| TRODELVY (n=268) |
단일 요법 화학 요법 (n=249) |
|||
|---|---|---|---|---|
| 이상 반응 | 모든 등급 % |
3-4등급 % |
모든 등급 % |
3-4등급 % |
| *단일 요법 화학 요법에는 다음 단일 약제 중 하나가 포함되었습니다. 에리불린(n=130), 비노렐빈(n=63), 젬시타빈(n=56) 또는 카페시타빈(n=22). | ||||
| NCI CTCAE v.5.0에 따라 등급이 매겨짐. | ||||
| 위장 장애 | ||||
| 설사 | 62 | 10 | 23 | 1 |
| 오심 | 59 | 1 | 35 | 3 |
| 변비 | 34 | 1 | 25 | 0 |
| 구토 | 23 | 1 | 16 | 2 |
| 복통 | 20 | 0 | 14 | 0 |
| 소화불량* | 11 | 0 | 6 | 0 |
| 일반 장애 및 투여 부위 상태 | ||||
| 피로† | 60 | 8 | 51 | 4 |
| 대사 및 영양 장애 | ||||
| 식욕 감퇴 | 21 | 2 | 21 | 0 |
| 저칼륨혈증 | 10 | 2 | 4 | 0 |
| 근골격 및 결합조직 장애 | ||||
| 관절통 | 15 | 0 | 12 | 0 |
| 신경계 장애 | ||||
| 두통 | 16 | 1 | 15 | 1 |
| 호흡, 흉부 및 종격동 장애 | ||||
| 호흡곤란 ‡ | 20 | 0 | 17 | 0 |
| 기침 | 12 | 0 | 7 | 0 |
| 피부 및 피하조직 장애 | ||||
| 탈모 | 48 | 0 | 19 | 0 |
| 가려움증 | 12 | 0 | 2 | 0 |
TROPiCS-02에서 임상적으로 유의미한 다른 이상반응 (≤ 10%)은 다음과 같습니다: 저혈압 (5%), 통증 (5%), 비루 (5%), 저칼슘혈증 (3%), 비충혈 (3%), 피부 색소침착 (3%), 결장염 또는 호중구감소증성 결장염 (2%), 저나트륨혈증 (2%), 폐렴 (2%), 단백뇨 (1%), 장염 (0.4%).
| 이상 실험실 검사 결과 | TRODELVY (n=268) |
단일 요법 항암화학요법 (n=249) |
||
|---|---|---|---|---|
| 모든 등급 (%) |
3-4등급 (%) |
모든 등급 (%) |
3-4등급 (%) |
|
| 혈액학 | ||||
| 백혈구 감소 | 88 | 38 | 73 | 26 |
| 호중구 감소 | 83 | 53 | 67 | 40 |
| 헤모글로빈 감소 | 73 | 8 | 59 | 5 |
| 림프구 감소 | 65 | 21 | 47 | 14 |
| 혈소판 감소 | 21 | 1 | 30 | 4 |
| 호산구 증가증 | 13 | 0 | 4 | 0 |
| 화학 | ||||
| 혈당 증가 | 37 | 0 | 31 | 0 |
| 알부민 감소 | 32 | 0 | 27 | 0.4 |
| 크레아티닌 청소율 감소 | 24 | 2 | 19 | 1 |
| 알칼리성 인산가수분해효소 증가 | 23 | 0 | 23 | 1 |
| 칼륨 감소 | 22 | 3 | 12 | 0.4 |
| 알라닌 아미노전이효소 증가 | 21 | 1 | 31 | 2 |
| 나트륨 감소 | 19 | 1 | 17 | 0.4 |
| 마그네슘 감소 | 18 | 0 | 15 | 0 |
| 인산염 감소 | 17 | 0 | 10 | 0 |
| 인산염 증가 | 16 | 0 | 16 | 0 |
| 젖산 탈수소효소 증가 | 16 | 0 | 28 | 0 |
| 아스파르트산 아미노전이효소 증가 | 15 | 2 | 25 | 1 |
| 칼륨 증가 | 14 | 2 | 9 | 0 |
7 약물 상호 작용
7.1 다른 약물이 TRODELVY에 미치는 영향
8 특정 집단에서의 사용
8.1 임신
위험 요약
작용 기전에 근거하여, TRODELVY는 임산부에게 투여될 경우 기형 유발 및/또는 배아-태아 치사를 유발할 수 있습니다. 약물 관련 위험을 알려주는 임산부에 대한 자료는 없습니다. TRODELVY는 유전독성 성분인 SN-38을 함유하고 있으며, 빠르게 분열하는 세포에 독성이 있습니다 [임상 약리학 (12.1) 및 비임상 독성학 (13.1) 참조]. 임신 여성과 임신 가능성이 있는 여성에게 태아에 대한 잠재적 위험을 알려주십시오.
해당 모집단에서 주요 선천적 기형 및 유산의 추정 배경 위험은 알려져 있지 않습니다. 미국 일반 인구에서 임상적으로 인지된 임신에서 주요 선천적 기형 및 유산의 추정 배경 위험은 각각 2~4% 및 15~20%입니다.
8.3 임신 가능성이 있는 여성 및 남성
피임
여성
TRODELVY는 임산부에게 투여될 경우 태아에게 해를 끼칠 수 있습니다 [특정 집단에서의 사용 (8.1) 참조]. 임신 가능성이 있는 여성에게 TRODELVY 치료 중 및 마지막 투여 후 6개월 동안 효과적인 피임법을 사용하도록 알려주십시오.
8.5 노인 사용
TRODELVY로 치료받은 삼중 음성 유방암 환자 366명 중 19%가 65세 이상이었고 3%가 75세 이상이었습니다. 65세 이상 환자와 그보다 젊은 환자 간에 안전성 및 유효성에 전반적인 차이는 관찰되지 않았습니다.
TRODELVY로 치료받은 HR+/HER2- 유방암 환자 322명 중 26%가 65세 이상이었고 6%가 75세 이상이었습니다. 65세 이상 환자와 그보다 젊은 환자 간에 유효성에 전반적인 차이는 관찰되지 않았습니다. 65세 이상 환자의 경우 유해 반응으로 인한 치료 중단율(14%)이 그보다 젊은 환자(3%)보다 높았습니다.
8.6 간 손상
경도 간 손상 환자에게 TRODELVY를 투여할 때 시작 용량을 조정할 필요가 없습니다 [임상 약리학 (12.3) 참조].
중등도(총 빌리루빈 > 1.5~3.0 × ULN) 또는 중증(총 빌리루빈 > 3.0 × 상한선 [ULN]) 간 손상 환자에서 TRODELVY의 안전성은 확립되지 않았습니다. TRODELVY는 간 전이가 없는 환자 또는 간 전이가 있는 환자에서 AST 또는 ALT > 3 ULN 또는 AST 또는 ALT > 5 ULN인 환자에게서 시험되지 않았습니다. 이러한 환자의 시작 용량에 대한 권장 사항은 없습니다.
10 과다 복용
임상 시험에서 최대 18 mg/kg(권장 최대 용량 10 mg/kg의 약 1.8배)의 TRODELVY 계획 용량이 투여되었습니다. 이러한 환자에서 중증 호중구 감소증의 발생률이 더 높았습니다.
11 설명
사시투주맙 고비테칸-hziy는 Trop-2 표적 항체-토포이소머라제 억제제 접합체로, 다음 세 가지 성분으로 구성됩니다.
- Trop-2 (영양막 세포 표면 항원-2)에 결합하는 인간화 단클론 항체 hRS7 IgG1κ (사시투주맙이라고도 함);
- 토포이소머라제 억제제인 SN-38;
- 인간화 단클론 항체를 SN-38에 연결하는 가수분해성 링커(CL2A라고 함).
재조합 단클론 항체는 포유류(마우스 골수종) 세포에 의해 생산되며, 소분자 성분 SN-38 및 CL2A는 화학 합성에 의해 생산됩니다. 사시투주맙 고비테칸-hziy는 항체 분자당 평균 7~8개의 SN-38 분자를 포함합니다. 사시투주맙 고비테칸-hziy의 분자량은 약 160킬로달톤입니다. 사시투주맙 고비테칸-hziy의 화학 구조는 다음과 같습니다.
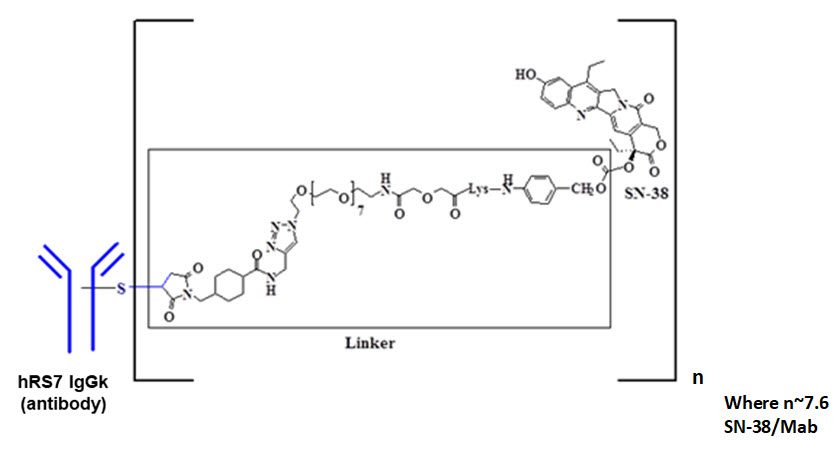 |
주사용 TRODELVY (사시투주맙 고비테칸-hziy)는 멸균 보존제가 없는, 연한 백색에서 황색을 띠는 동결건조 분말로, 정맥 주사용 50mL 투명 유리 1회용 바이알에 고무 마개가 있으며 알루미늄 캡으로 밀봉되어 있습니다.
TRODELVY 1회용 바이알에는 사시투주맙 고비테칸-hziy 180mg, 2-(N-모르폴리노)에탄술폰산(MES) 71.7mg, 폴리소르베이트 80 1.8mg, 트레할로스 153.99mg이 들어 있습니다. 20mL의 0.9% 염화나트륨 주사액 USP로 재구성하면 농도는 10mg/mL이고 pH는 6.5가 됩니다.
12 약물동력학
12.1 작용 기전
사시투주맙 고비테칸-hziy는 Trop-2 표적 항체-약물 접합체입니다. 사시투주맙은 Trop-2를 인식하는 인간화 항체입니다. 소분자 SN-38은 토포이소머라제 I 억제제이며, 링커를 통해 항체에 공유 결합되어 있습니다. 약리학적 데이터에 따르면 사시투주맙 고비테칸-hziy는 Trop-2를 발현하는 암세포에 결합하고, 이후 링커의 가수분해를 통해 SN-38이 방출되면서 세포 내로 유입됩니다. SN-38은 토포이소머라제 I와 상호 작용하여 토포이소머라제 I에 의해 유도된 단일 가닥 절단의 재결합을 방지합니다. 그 결과 DNA 손상이 발생하여 세포자멸사 및 세포 사멸이 일어납니다. 사시투주맙 고비테칸-hziy는 삼중 음성 유방암의 마우스 이종이식 모델에서 종양 성장을 감소시켰습니다.
12.3 약동학
10 mg/kg의 단일 요법으로 사시투주맙 고비테칸-hziy를 투여받은 mBC 환자에서 사시투주맙 고비테칸-hziy와 SN-38의 혈청 약동학이 평가되었습니다. 사시투주맙 고비테칸-hziy와 유리 SN-38의 약동학적 매개변수는 표 8에 제시되어 있습니다.
| 사시투주맙 고비테칸-hziy (N=693) |
유리 SN-38 (N=681) |
|
|---|---|---|
| Cmax: 첫 번째 투여 후 0~168시간 동안의 최대 혈청 농도 | ||
| AUC0–168: 첫 번째 투여 후 168시간까지의 혈청 농도 곡선 아래 면적 | ||
|
||
| Cmax [ng/mL] | 239000 (11%) | 98.0 (45%) |
| AUC0–168 [ng*h/mL] | 5640000 (22%) | 3696 (56%) |
제거
전이성 삼중 음성 유방암 환자에서 sacituzumab govitecan-hziy와 유리 SN-38의 중앙 제거 반감기(t1/2)는 각각 23.4시간과 17.6시간이었습니다. 모집단 약동학 분석에 따르면, sacituzumab govitecan-hziy의 추정 평균(%CV) 청소율은 0.13 L/h (12%)입니다.
대사
sacituzumab govitecan-hziy에 대한 대사 연구는 수행되지 않았습니다. SN-38 (sacituzumab govitecan-hziy의 소분자 부분)은 UGT1A1을 통해 대사됩니다. SN-38의 글루쿠로나이드 대사체(SN-38G)는 환자의 혈청에서 검출되었습니다.
특정 집단
TRODELVY로 치료받은 환자에 대한 약동학 분석 결과, 연령(27~88세), 인종(백인, 흑인 또는 아시아인) 또는 경도 신장애에서 중등도 신장애(CLcr 30~89 mL/min)가 sacituzumab govitecan-hziy의 약동학에 미치는 영향은 확인되지 않았습니다. 신장 배설은 sacituzumab govitecan-hziy의 소분자 부분인 SN-38의 배설에 최소한으로 기여하는 것으로 알려져 있습니다. 중증 신장애(CLcr 15~29 mL/min) 또는 말기 신질환(CLcr < 15 mL/min) 환자에 대한 sacituzumab govitecan-hziy의 약동학 데이터는 없습니다.
간 손상 환자
경도 간 손상 환자(총 빌리루빈 ≤ ULN, AST > ULN 또는 빌리루빈 >1.0~≤ 1.5 ULN, AST 임의; n=257)에서 sacituzumab govitecan-hziy의 노출은 정상 간 기능 환자(총 빌리루빈 또는 AST < ULN; n=526)와 유사합니다.
중등도(총 빌리루빈 > 1.5~3.0 × ULN) 또는 중증(총 빌리루빈 > 3.0 × ULN) 간 손상 환자에서 sacituzumab govitecan-hziy와 유리 SN-38의 노출은 알려져 있지 않습니다.
약물 상호작용 연구
sacituzumab govitecan-hziy 또는 그 구성 요소에 대한 약물 상호작용 연구는 수행되지 않았습니다. UGT1A1 억제제 또는 유도제는 각각 SN-38 노출을 증가시키거나 감소시킬 수 있습니다 [약물 상호작용 (7) 참조].
12.5 약물 유전체학
SN-38은 UGT1A1을 통해 대사됩니다 [임상 약리학 (12.3) 참조]. UGT1A1*28 대립 유전자와 같은 UGT1A1 유전자의 유전적 변이는 UGT1A1 효소 활성을 감소시킵니다. UGT1A1*28 대립 유전자에 대해 동형 접합 또는 이형 접합인 개인은 야생형(*1/*1) 개인에 비해 TRODELVY로 인한 호중구 감소증, 발열성 호중구 감소증 및 빈혈 위험이 증가합니다 [경고 및 주의사항 (5.5) 참조]. 흑인 또는 아프리카계 미국인 인구의 약 20%, 백인 인구의 약 10%, 동아시아 인구의 약 2%가 UGT1A1*28 대립 유전자(*28/*28)에 대해 동형 접합입니다. 흑인 또는 아프리카계 미국인 인구의 약 40%, 백인 인구의 약 50%, 동아시아 인구의 약 25%가 UGT1A1*28 대립 유전자(*1/*28)에 대해 이형 접합입니다. UGT1A1*28 이외의 감소 기능 대립 유전자는 특정 인구 집단에 존재할 수 있습니다.
12.6 면역원성
항체 형성의 검출은 분석법의 민감도와 특이성에 크게 의존합니다. 분석 방법의 차이로 인해 아래에 설명된 연구에서 항체의 발생률과 다른 연구(TRODELVY 연구 포함)에서 항체의 발생률을 의미 있게 비교할 수 없습니다.
TRODELVY로 치료받은 환자에 대한 임상 연구에서 중앙값 4개월 치료 기간 동안 785명의 환자 중 9명(1.1%)이 sacituzumab govitecan에 대한 항체를 생성했습니다. 이 환자 중 6명(TRODELVY로 치료받은 모든 환자의 0.8%)은 sacituzumab govitecan에 대한 중화 항체를 가지고 있었습니다. 항체 발생률이 낮기 때문에 이러한 항체가 sacituzumab govitecan의 약동학, 약력학, 안전성 및/또는 효과에 미치는 영향은 알려져 있지 않습니다.
13 비임상 독성학
13.1 발암성, 돌연변이원성, 생식능력 저하
사시투주맙 고비테칸-hziy를 사용한 발암성 연구는 수행되지 않았습니다.
SN-38은 중국 햄스터 난소 세포에서 in vitro 포유류 세포 소핵 시험에서 클라스토젠성을 나타냈으며, in vitro 박테리아 역돌연변이 (Ames) 분석에서는 돌연변이원성을 나타내지 않았습니다.
사시투주맙 고비테칸-hziy를 사용한 생식능력 연구는 수행되지 않았습니다. 시노몰구스 원숭이를 대상으로 한 반복 투여 독성 연구에서, 1일차와 4일차에 사시투주맙 고비테칸-hziy를 정맥 투여한 결과, 60mg/kg 이상(체중 기준 10mg/kg의 사람 권장 용량의 6배 이상)의 용량에서 자궁내막 위축, 자궁 출혈, 난소의 여포 폐쇄 증가 및 질 상피 세포 위축이 발생했습니다.
14 CLINICAL STUDIES
14.1 국소 진행성 또는 전이성 삼중 음성 유방암
ASCENT
유방암에 대해 최소 두 가지 이상의 이전 화학요법(신보조요법 또는 보조요법 환경에서 하나일 수 있음, 단 진행이 12개월 이내에 발생한 경우)을 받은 후 재발한 절제 불가능한 국소 진행성 또는 전이성 삼중 음성 유방암(mTNBC) 환자 529명을 대상으로 다기관, 공개, 무작위 연구(ASCENT; NCT02574455)에서 효능을 평가했습니다. 첫 번째 택산 사이클 중 또는 끝날 무렵 택산에 대한 금기 사항이나 불내성이 없는 한 모든 환자는 보조요법, 신보조요법 또는 진행 단계에서 이전 택산 치료를 받았습니다. 알려진 또는 의심되는 뇌 전이가 있는 환자의 경우 등록 전에 뇌 전이를 확인하기 위한 자기공명영상(MRI)이 필요했습니다. 뇌 전이가 있는 환자는 ASCENT 연구에서 미리 정의된 최대 15%까지 등록할 수 있었습니다. 알려진 길버트 증후군 또는 뼈 질환만 있는 환자는 제외되었습니다.
환자는 21일마다 1일차와 8일차에 정맥 주입으로 TRODELVY 10mg/kg을 투여받는 그룹(n=267) 또는 의사가 선택한 단일 요법 화학요법(n=262)을 투여받는 그룹으로 1:1의 비율로 무작위 배정되었습니다. 단일 요법 화학요법은 무작위 배정 전에 연구자가 다음 중 하나를 선택하여 결정했습니다. 에리불린(n=139), 카페시타빈(n=33), 젬시타빈(n=38), 또는 비노렐빈(n=52).
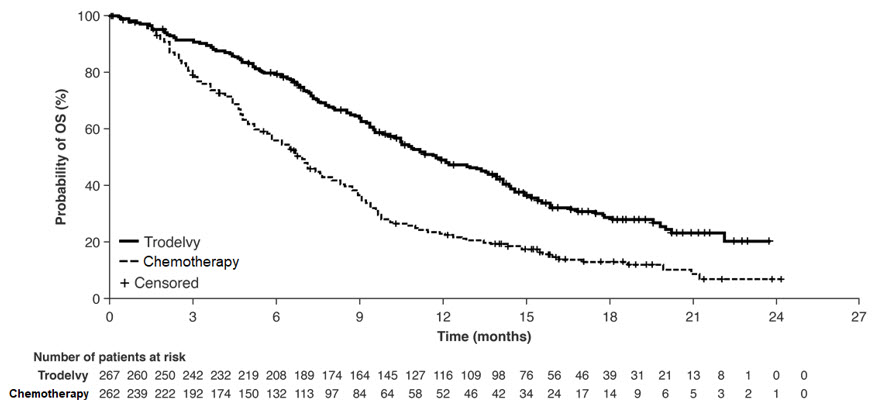
환자는 질병 진행 또는 허용할 수 없는 독성이 나타날 때까지 치료를 받았습니다. 주요 효능 결과는 반응 평가 기준 고형 종양(RECIST) v1.1 기준을 사용하여 맹검, 독립적, 중앙 집중 검토를 통해 측정한 기준 시점에 뇌 전이가 없는 환자(즉, BMNeg)의 무진행 생존 기간(PFS)이었습니다. 추가 효능 측정에는 전체 환자군(뇌 전이가 있는 환자와 없는 환자 모두)의 PFS 및 전체 생존 기간(OS)이 포함되었습니다.
전체 환자군(n=529)의 중앙 연령은 54세(범위: 27~82세)였으며, 99.6%가 여성이었고, 79%가 백인, 12%가 흑인/아프리카계 미국인이었으며, 81%의 환자가 65세 미만이었습니다. 모든 환자의 ECOG 수행 상태는 0(43%) 또는 1(57%)이었습니다. 42%의 환자에게 간 전이가 있었고, 9%는 BRCA1/BRCA2 돌연변이 상태가 양성이었으며, 70%는 진단 시 TNBC였습니다. 12%는 이전에 치료받고 안정적인 기준 시점 뇌 전이가 있었습니다(n=61; TRODELVY군 32명, 단일 요법 화학요법군 29명). 전반적으로 29%의 환자가 이전 PD-1/PD-L1 치료를 받았습니다. 전체 환자군 중 TRODELVY군의 13%는 전이 환경에서 이전 전신 치료를 1회만 받았습니다.
효능 결과는 표 9에 요약되어 있으며 그림 1과 그림 2에 나와 있습니다. 전이 환경에서 이전 전신 치료를 1회만 받은 환자 하위 그룹(신보조요법/보조요법 전신 치료 후 12개월 이내에 질병 재발 또는 진행이 있는 경우)의 효능 결과는 전이 환경에서 최소 두 가지 이상의 치료를 받은 환자의 결과와 일치했습니다.
| 무작위 배정된 모든 환자 | ||
|---|---|---|
| TRODELVY n=267 |
단일 요법 화학요법 n=262 |
|
| CI = 신뢰 구간 | ||
| BICR에 따른 무진행 생존 기간* | ||
| 질병 진행 또는 사망 (%) | 190 (71%) | 171 (65%) |
| 월 단위 중앙 PFS (95% CI) | 4.8 (4.1, 5.8) |
1.7 (1.5, 2.5) |
| 위험 비† (95% CI) | 0.43 (0.35, 0.54) | |
| p-값 | <0.0001 | |
| 전체 생존 기간 | ||
| 사망 (%) | 179 (67%) | 206 (79%) |
| 월 단위 중앙 OS (95% CI) | 11.8 (10.5, 13.8) |
6.9 (5.9, 7.6) |
| 위험 비† (95% CI) | 0.51 (0.41, 0.62) | |
| p-값 | <0.0001 | |
그림 1: ASCENT 연구에서 BICR에 의한 무진행 생존율의 Kaplan-Meier 곡선 (모든 무작위 배정 환자)
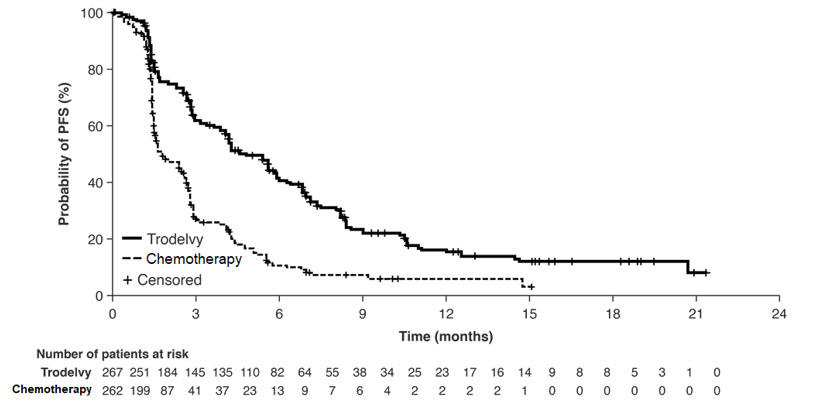
그림 2: ASCENT 연구에서 전체 생존율의 Kaplan-Meier 곡선 (모든 무작위 배정 환자)
이전에 치료받은 안정적인 뇌 전이 환자의 무진행 생존율에 대한 탐색적 분석 결과, 계층화된 위험비는 0.65 (95% CI: 0.35, 1.22)였습니다. TRODELVY군의 무진행 생존율 중앙값은 2.8개월 (95% CI: 1.5, 3.9)이었고, 단독 화학요법군의 무진행 생존율 중앙값은 1.6개월 (95% CI: 1.3, 2.9)이었습니다. 동일한 집단에서의 탐색적 전체 생존율 분석 결과, 계층화된 위험비는 0.87 (95% CI: 0.47, 1.63)이었습니다. TRODELVY군의 전체 생존율 중앙값은 6.8개월 (95% CI: 4.7, 14.1)이었고, 단독 화학요법군의 전체 생존율 중앙값은 7.4개월 (95% CI: 4.7, 11.1)이었습니다.
IMMU-132-01
TRODELVY의 효능은 다기관 단일군 연구 (NCT01631552)에서 평가되었으며, 이 연구에는 전이성 질환에 대해 2회 이상의 항암 치료를 받은 전이성 삼중 음성 유방암 (mTNBC) 환자 108명이 등록되었습니다. 종괴 크기가 7cm 초과인 환자는 연구 대상에서 제외되었습니다. 고용량 스테로이드 (프레드니손 20mg 이상 또는 동등 약물)를 4주 이상 복용하지 않은 뇌 전이 환자는 연구 대상에 포함되었습니다. 길버트 증후군이 있는 환자는 제외되었습니다.
환자들은 21일 치료 주기의 1일차와 8일차에 정맥 내로 TRODELVY 10 mg/kg을 투여받았습니다. 환자들은 질병 진행 또는 치료에 대한 불내성이 나타날 때까지 TRODELVY로 치료받았습니다. 종양 영상 검사는 8주마다 시행되었으며, 초기 부분 반응 또는 완전 반응 후 4~6주 후에 확인을 위한 CT/MRI 검사를 시행하여 치료 중단이 필요한 진행이 나타날 때까지 계속되었습니다. 주요 효능 결과 측정 항목은 RECIST 1.1을 사용한 연구자 평가 전체 반응률 (ORR) 및 반응 지속 기간이었습니다.
중앙값 연령은 55세 (범위: 31~80세)였으며, 87%의 환자가 65세 미만이었습니다. 대부분의 환자는 여성 (99%)이었고 백인 (76%)이었습니다. 연구 시작 시 모든 환자의 ECOG 성능 상태는 0 (29%) 또는 1 (71%)이었습니다. 76%의 환자가 내장 질환을 가지고 있었고, 42%는 간 전이, 56%는 폐/흉막 전이, 2%는 뇌 전이를 가지고 있었습니다. 12명의 환자 (11%)가 초기 진단 당시 4기 질환을 가지고 있었습니다.
전이 환경에서 이전에 받은 전신 치료의 중앙값은 3회 (범위: 2~10회)였습니다. 전이 환경에서 이전에 받은 화학 요법에는 카보플라틴 또는 시스플라틴 (69%), 젬시타빈 (55%), 팍리탁셀 또는 도세탁셀 (53%), 카페시타빈 (51%), 에리불린 (45%), 독소루비신 (24%), 비노렐빈 (16%), 사이클로포스파마이드 (19%) 및 익사베필론 (8%)이 포함되었습니다.
전반적으로 98%의 환자가 이전에 택산을, 86%의 환자가 (신보조 또는 전이 환경에서) 이전에 안트라사이클린을 투여받았습니다.
표 10은 효능 결과를 요약한 것입니다.
14.2 국소 진행성 또는 전이성 HR-양성, HER2-음성 유방암
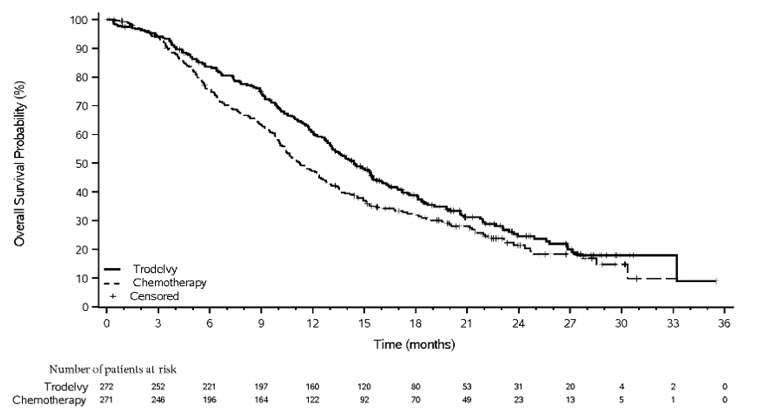
TROPiCS-02 연구
TRODELVY의 효능은 국소 진행성 또는 전이성 HR-양성, HER2-음성 (IHC 0, IHC 1+ 또는 IHC 2+/ISH–) 유방암 환자 543명을 대상으로 다기관, 공개, 무작위 연구(TROPiCS-02; NCT03901339)에서 평가되었습니다. 이 환자들은 어떤 치료 환경에서든 다음 치료 후 질병이 진행된 경우였습니다. CDK 4/6 억제제, 내분비 요법 및 택산; 환자들은 전이 환경에서 최소 두 가지 이상의 이전 화학요법(신보조 또는 보조 환경에서 하나일 수 있음, 재발이 12개월 이내에 발생한 경우)을 받았습니다.
환자들은 21일 주기의 1일차와 8일차에 정맥 주입으로 TRODELVY 10mg/kg을 투여받는 그룹(n=272) 또는 단일 요법 화학요법을 투여받는 그룹(n=271)에 1:1로 무작위 배정되었습니다. 단일 요법 화학요법은 무작위 배정 전 연구자가 다음 중 하나를 선택하여 결정했습니다. 에리불린(n=130), 비노렐빈(n=63), 젬시타빈(n=56) 또는 카페시타빈(n=22). 무작위 배정은 다음 요인에 따라 계층화되었습니다. 전이 질환에 대한 이전 화학요법 요법(2대 3-4), 내장 전이(예 또는 아니오) 및 최소 6개월 동안 전이 환경에서의 내분비 요법(예 또는 아니오).
환자들은 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 치료를 받았습니다. 환자가 임상적으로 안정적이고 연구자가 임상적 이점을 얻고 있다고 판단한 경우 RECIST로 정의된 질병 진행을 넘어 TRODELVY 투여가 허용되었습니다. 1차 유효성 결과 측정은 RECIST v1.1에 따라 BICR에 의해 결정된 PFS였습니다. 추가적인 유효성 측정에는 OS, BICR에 의한 ORR 및 BICR에 의한 DOR이 포함되었습니다.
연구 대상 환자의 중앙값 연령은 56세(범위: 27–86세)였으며, 26%의 환자가 65세 이상이었습니다. 대부분의 환자는 여성(99%)이었으며, 67%는 백인, 4%는 흑인, 3%는 아시아인이었고 26%는 인종이 알려지지 않았습니다. 환자들은 어떤 환경에서든 중앙값 7개(범위: 3~17개)의 이전 전신 요법과 전이 환경에서 중앙값 3개(범위: 0~8개)의 이전 전신 화학요법 요법을 받았습니다. 약 42%의 환자가 전이 질환 치료를 위한 2가지 이전 화학요법 요법을 받은 반면, 58%의 환자는 3~4가지 이전 화학요법 요법을 받았으며 모든 환자의 ECOG 수행 상태는 0(45%) 또는 1(55%)이었습니다. 95%의 환자에게 내장 전이가 있었습니다. 대부분의 환자는 ≥ 6개월 동안 전이 환경에서 내분비 요법을 받았습니다(86%).
TRODELVY는 단일 요법 화학요법에 비해 PFS 및 OS에서 통계적으로 유의미한 개선을 보였습니다.
유효성 결과는 표 11과 그림 3 및 그림 4에 요약되어 있습니다.
| 모든 무작위 배정 환자 | ||
|---|---|---|
| TRODELVY n=272 |
단일 요법 화학요법 n=271 |
|
| BICR에 의한 무진행 생존기간* | ||
| 월 단위 중앙값 PFS (95% CI) |
5.5 (4.2, 7.0) |
4.0 (3.1, 4.4) |
| 위험 비(95% CI) | 0.661 (0.529, 0.826) | |
| p-값† | 0.0003 | |
| 전체 생존기간‡ | ||
| 월 단위 중앙값 OS (95% CI) |
14.4 (13.0, 15.7) |
11.2 (10.1, 12.7) |
| 위험 비(95% CI) | 0.789 (0.646, 0.964) | |
| p-값† | 0.0200 | |
| BICR에 의한 객관적 반응률 | ||
| 반응률, %(95% CI) | 21.0 (16.3, 26.3) | 14.0 (10.1, 18.7) |
| 오즈비(95% CI) | 1.625 (1.034, 2.555) | |
| p-값 | 0.0348 | |
| BICR에 의한 반응 지속 기간(DOR) | ||
| 중앙 DOR (월) (95% CI) |
8.1 (6.7, 9.1) |
5.6 (3.8, 7.9) |
그림 3: TROPiCS-02에서 BICR에 따른 PFS의 Kaplan-Meier 플롯
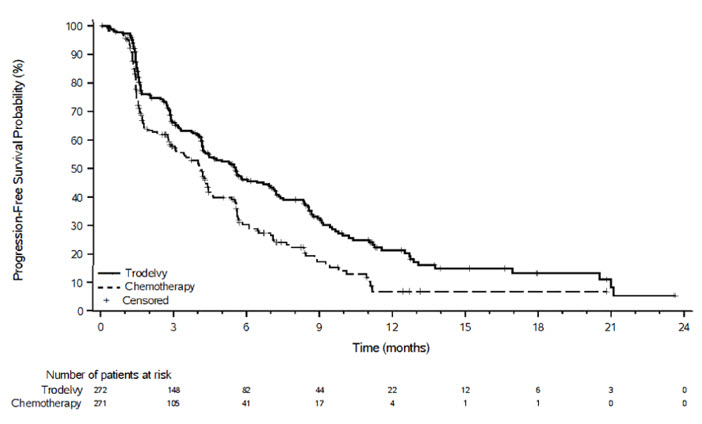
그림 4: TROPiCS-02에서 OS의 Kaplan-Meier 플롯
15 참고 문헌
1. “OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html. (OSHA 위험 약물. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.)
16 제공/보관 및 취급 방법
TRODELVY (sacituzumab govitecan-hziy) 주사용은 멸균된 회백색~황백색의 동결건조 분말로 단회 투여용 바이알에 담겨 있습니다. 각 TRODELVY 바이알은 개별적으로 상자에 포장되어 있습니다.
- NDC 55135-132-01 에는 180mg 바이알 1개가 들어 있습니다.
재구성할 때까지 바이알을 원래 상자에 넣어 2°C~8°C (36°F~46°F)의 냉장고에 보관하여 빛으로부터 보호하십시오. 얼리지 마십시오.
TRODELVY는 위험한 약물입니다. 해당되는 특별 취급 및 폐기 절차를 따르십시오1.
17 환자 상담 정보
FDA 승인 환자용 설명서(Patient Information)를 읽도록 환자에게 알려주십시오.
호중구 감소증
환자에게 호중구 감소증의 위험성을 알려주십시오. 환자에게 발열, 오한 또는 기타 감염 징후가 나타나면 즉시 의료 제공자에게 연락하도록 지시하십시오 [경고 및 주의 사항 (5.1) 참조].
설사
환자에게 설사의 위험성을 알려주십시오. 환자에게 치료 중 처음으로 설사가 발생하거나, 검은색 또는 피가 섞인 변, 어지러움, 현기증 또는 실신과 같은 탈수 증상, 메스꺼움이나 구토로 인해 경구 섭취가 불가능하거나, 24시간 이내에 설사를 조절할 수 없는 경우 즉시 의료 제공자에게 연락하도록 지시하십시오 [경고 및 주의 사항 (5.2) 참조].
과민반응 및 주입 관련 반응
환자에게 심각한 주입 반응 및 아나필락시스의 위험성을 알려주십시오. 환자에게 얼굴, 입술, 혀 또는 목의 부종, 두드러기, 호흡 곤란, 어지러움, 현기증, 오한, 오한, 쌕쌕거림, 가려움증, 홍조, 발진, 저혈압 또는 주입 중 또는 주입 후 24시간 이내에 발생하는 발열이 나타나면 즉시 의료 제공자에게 연락하도록 지시하십시오 [경고 및 주의 사항 (5.3) 참조].
메스꺼움/구토
환자에게 메스꺼움과 구토의 위험성을 알려주십시오. 항암 화학 요법 유발 메스꺼움 및 구토(CINV) 예방을 위한 2가지 또는 3가지 약물 요법으로 확립된 지침에 따른 전처치도 권장됩니다. 임상적으로 필요한 경우 추가 항구토제, 진정제 및 기타 보조 치료법을 사용할 수도 있습니다. 모든 환자는 지연된 메스꺼움과 구토를 예방하고 치료하기 위한 처방약을 명확한 지침과 함께 받아야 합니다. 환자에게 조절되지 않는 메스꺼움이나 구토가 발생하면 즉시 의료 제공자에게 연락하도록 지시하십시오 [경고 및 주의 사항 (5.4) 참조].
태아-태아 독성
여성 환자에게 임신 중이거나 임신하게 되면 의료 제공자에게 연락하도록 알려주십시오. 여성 환자에게 태아에 대한 위험과 임신 손실 가능성을 알려주십시오 [특정 집단에서의 사용 (8.1) 참조].
피임
생식 능력이 있는 여성 환자에게 TRODELVY 치료 중 및 마지막 투여 후 6개월 동안 효과적인 피임법을 사용하도록 알려주십시오 [특정 집단에서의 사용 (8.3) 참조].
생식 능력이 있는 여성 파트너가 있는 남성 환자에게 TRODELVY 치료 중 및 마지막 투여 후 3개월 동안 효과적인 피임법을 사용하도록 알려주십시오 [특정 집단에서의 사용 (8.3) 참조].
SPL 미분류 섹션
제조원:
Gilead Sciences, Inc.
333 Lakeside Dr.
Foster City, CA 94404, USA
미국 허가 번호 2258
환자 사용 설명서
| 본 환자 정보는 미국 식품의약국(FDA)의 승인을 받았습니다. | 개정: 2024년 11월 | |
| 환자 정보 TRODELVY® (트로델비) (sacituzumab govitecan-hziy) 주사제, 정맥 투여용 |
||
|
TRODELVY에 대해 가장 중요한 정보는 무엇입니까?
|
||
|
|
|
|
||
| TRODELVY란 무엇입니까? TRODELVY는 다음과 같은 성인 환자를 치료하는 처방약입니다.
중등도 또는 중증 간 문제가 있는 사람에게 TRODELVY가 안전하고 효과적인지 여부는 알려져 있지 않습니다. |
||
| TRODELVY에 대한 심각한 알레르기 반응이 있었던 경우 TRODELVY를 투여받지 마십시오. 확실하지 않은 경우 의료진에게 문의하십시오. | ||
TRODELVY를 투여받기 전에 다음과 같은 경우를 포함하여 모든 의학적 상태에 대해 의료진에게 알리십시오.
처방약과 일반의약품, 비타민 및 허브 보충제를 포함하여 복용하는 모든 약에 대해 의료진에게 알리십시오. 특정 약물은 TRODELVY의 작용 방식에 영향을 줄 수 있습니다. |
||
TRODELVY는 어떻게 투여받게 됩니까?
|
||
| TRODELVY의 가능한 부작용은 무엇입니까? TRODELVY는 다음을 포함한 심각한 부작용을 유발할 수 있습니다.
|
||
|
|
|
TRODELVY의 가장 흔한 부작용은 다음과 같습니다. |
||
|
|
|
| TRODELVY는 여성의 생식 능력에 문제를 일으켜 임신 능력에 영향을 미칠 수 있습니다. 생식 능력이 걱정되는 경우 의료 서비스 제공자와 상담하십시오. 이것들은 TRODELVY의 모든 가능한 부작용이 아닙니다. 부작용에 대한 의학적 조언은 의사에게 문의하십시오. FDA에 부작용을 보고할 수 있습니다(1-800-FDA-1088). |
||
| TRODELVY의 안전하고 효과적인 사용에 대한 일반 정보. 의약품은 때때로 환자 정보 설명서에 나열된 목적 이외의 목적으로 처방되기도 합니다. 의료 전문가를 위해 작성된 TRODELVY에 대한 정보는 약사 또는 의료 서비스 제공자에게 문의할 수 있습니다. |
||
| TRODELVY의 성분은 무엇입니까? 활성 성분: sacituzumab govitecan-hziy 비활성 성분: 2-(N-morpholino) ethane sulfonic acid (MES), polysorbate 80 및 trehalose 제조사: Gilead Sciences, Inc., 333 Lakeside Dr., Foster City, CA 94404, USA 미국 허가 번호 2258 761115-GS-009 TRODELVY에 대한 자세한 내용은 www.TRODELVY.com을 방문하거나 1-888-983-4668로 전화하십시오. |
||
주요 표시 패널 – 180mg 바이알 라벨
NDC 55135-132-01
처방전 의약품
TRODELVY®
sacituzumab govitecan-hziy
주사용
바이알당 180 mg
정맥 주입 전용
경고: 위험 의약품
1회용 바이알
사용하지 않은 부분은 버리십시오
90370103

주요 표시 패널 – 180mg 바이알 상자
NDC 55135-132-01
처방전 의약품
TRODELVY®
sacituzumab govitecan-hziy
주사용
바이알당 180 mg
정맥 주입 전용
경고: 위험 의약품
사용 직전에 즉시 재구성 및 희석하십시오
1회용 바이알
사용하지 않은 부분은 버리십시오
1 바이알
