
의약품 제조업체: Clovis Oncology, Inc. (Updated: 2024-02-21)
처방 정보 요약
RUBRACA
®(루카파립) 정제, 경구용
미국 최초 승인: 2016
최근 주요 변경 사항
적응증 및 사용법
RUBRACA는 다음과 같은 경우에 사용되는 폴리(ADP-리보스) 폴리머라제(PARP) 억제제입니다.
난소암
- 백금 기반 화학 요법에 대한 완전 또는 부분 반응을 보이는 유전자 및/또는 체세포
BRCA 돌연변이(손상)와 관련된 재발성 상피성 난소암, 난관암 또는 원발성 복막암이 있는 성인 환자의 유지 치료. (
1.1)
전립선암
- 안드로겐 수용체 표적 치료 및 택산 기반 화학 요법을 받은 유전자 및/또는 체세포
BRCA 돌연변이(손상)와 관련된 전이성 거세 저항성 전립선암(mCRPC)이 있는 성인 환자의 치료. RUBRACA에 대한 FDA 승인 동반 진단을 기반으로 치료를 위한 환자를 선별하십시오. (
1.2,
2.1)
이 적응증은 객관적 반응률 및 반응 지속 기간을 기반으로 한 가속 승인에 따라 승인되었습니다. 이 적응증에 대한 지속적인 승인은 확인 시험에서 임상적 이점의 검증 및 설명에 따라 달라질 수 있습니다. (
1.2)
투여량 및 투여 방법
투여 형태 및 강도
정제: 200mg, 250mg 및 300mg (
3)
금기 사항
없음. (
4)
경고 및 주의 사항
부작용
- 난소암 환자에서 가장 흔한 부작용(≥ 10%)은 메스꺼움, 피로(무력증 포함), 빈혈, AST/ALT 증가, 구토, 설사, 식욕 감소, 혈소판 감소증, 미각 이상, 호중구 감소증, 혈중 크레아티닌 증가, 호흡 곤란, 현기증, 소화 불량, 광과민 반응 및 백혈구 감소증이었습니다. (
6.1)
-
BRCA 돌연변이 mCRPC 환자에서 가장 흔한 부작용(≥ 20%)은 피로(무력증 포함), 메스꺼움, 빈혈, ALT/AST 증가, 식욕 감소, 발진, 변비, 혈소판 감소증, 구토, 설사였습니다. (
6.1)
의심되는 부작용을 보고하려면 Clovis Oncology, Inc.에 1-844-258-7662로 연락하거나 FDA에 1-800-FDA-1088로 연락하거나www.fda.gov/medwatch를 방문하십시오.
환자 상담 정보 및 FDA 승인 환자 라벨은 17을 참조하십시오.
개정: 12/2022
목차
전문 정보: 내용*
1 적응증 및 사용법
1.1
BRCA 돌연변이 재발성 난소암의 유지 요법
1.2
BRCA 돌연변이 전이성 거세 저항성 전립선암
2 용법 및 용량
2.1 환자 선택
2.2 권장 용량
2.3 이상 반응에 대한 용량 조절
3 제형 및 강도
4 금기 사항
5 경고 및 주의 사항
5.1 골수이형성증후군/급성 골수성 백혈병
5.2 태아 독성
6 이상 반응
6.1 임상 시험 경험
7 약물 상호 작용
7.1 Rubraca의 다른 약물에 대한 영향
8 특정 인구 집단에서의 사용
8.1 임신
8.2 수유
8.3 생식 능력이 있는 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 신장애
8.7 간장애
11 설명
12 약리학
12.1 작용 기전
12.2 약력학
12.3 약동학
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식 능력 저해
14 임상 연구
14.1
BRCA 돌연변이 재발성 난소암의 유지 요법
14.2
BRCA 돌연변이 전이성 거세 저항성 전립선암
16 포장 단위/보관 및 취급
17 환자 상담 정보
- *
- 전문 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1 적응증 및 용법
1.1
BRCA 돌연변이 재발성 난소암의 유지 요법
Rubraca는 백금 기반 화학 요법에 대한 완전 또는 부분 반응을 보이는 성인 환자에서 유전자 및/또는 체세포
BRCA 돌연변이(deleterious)와 관련된 재발성 상피성 난소암, 난관암 또는 원발성 복막암의 유지 요법에 사용됩니다.
1.2
BRCA 돌연변이 전이성 거세 저항성 전립선암
Rubraca는 안드로겐 수용체 표적 치료 및 택산 기반 화학 요법을 받은 성인 환자에서 유전자 및/또는 체세포
BRCA 돌연변이(deleterious)와 관련된 전이성 거세 저항성 전립선암(mCRPC)의 치료에 사용됩니다. Rubraca에 대한 FDA 승인 동반 진단을 기반으로 치료 환자를 선별하십시오.
[용법 및 투여(
2.1) 참조].
이 적응증은 객관적 반응률 및 반응 지속 기간을 기반으로 한 가속 승인에 따라 승인되었습니다.
[임상 연구(
14.2) 참조]
. 이 적응증에 대한 지속적인 승인은 확증적 시험에서 임상적 이점의 검증 및 설명에 따라 달라질 수 있습니다.
2 용법 및 용량
2.1 환자 선택
재발성 난소암의 유지 치료BRCA 돌연변이
Rubraca를 이용한 재발성 난소암의 유지 치료를 위해서는 유해한
BRCA돌연변이(생식세포 및/또는 체세포)가 있는 환자를 선택해야 합니다 [
임상 연구(
14.1)
참조].
현재 유해한 생식세포 및/또는 체세포
BRCA돌연변이를 검출하는 FDA 승인 검사는 제공되지 않습니다.
치료BRCA 돌연변이 mCRPC (안드로겐 수용체 표적 치료 및 화학 요법 후)
Rubraca를 이용한 mCRPC 치료를 위해서는 혈장 검체에서 유해한
BRCA돌연변이(생식세포 및/또는 체세포)가 있는 환자를 선택해야 합니다
[임상 연구(
14.2) 참조].
혈장 검체에서 음성 결과가 나왔다고 해서 환자의 종양에
BRCA돌연변이가 없다는 것을 의미하지는 않습니다. 혈장 검체에서 음성 결과가 나온 경우 임상적으로 적절하다면 종양 검체를 이용하여 추가적인 유전체 검사를 고려해야 합니다.
난소암 또는 전립선암 환자의
BRCA돌연변이 검출을 위한 FDA 승인 검사에 대한 정보는 다음 웹사이트에서 확인할 수 있습니다. http://www.fda.gov/CompanionDiagnostics.
2.2 권장 용량
Rubraca의 권장 용량은 1일 2회, 음식과 함께 또는 음식 없이 경구로 600mg(300mg 정제 2정)을 복용하여 총 1일 용량 1,200mg을 복용하는 것입니다.
질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 치료를 계속합니다.
환자가 Rubraca 복용량을 놓친 경우, 다음 복용량을 예정된 시간에 복용하도록 지시합니다. 구토로 인해 복용량을 놓친 경우에는 다시 복용하지 않습니다.
mCRPC에 대해 Rubraca를 복용하는 환자는 동시에 성선 자극 호르몬 방출 호르몬(GnRH) 유사체를 투여하거나 양측 고환 절제술을 시행해야 합니다.
2.3 부작용에 대한 용량 조절
부작용을 관리하기 위해 치료 중단 또는 용량 감소를 고려하십시오. 부작용에 대한 권장 Rubraca 용량 조절은
표 1에 나와 있습니다.
| 용량 감소 | 용량 |
| 시작 용량 | 1일 2회 600mg(300mg 정제 2정) |
| 첫 번째 용량 감소 | 1일 2회 500mg(250mg 정제 2정) |
| 두 번째 용량 감소 | 1일 2회 400mg(200mg 정제 2정) |
| 세 번째 용량 감소 | 1일 2회 300mg(300mg 정제 1정) |
3. 투여 형태 및 강도
- 정제 (200 mg): 파란색, 둥근 모양, 즉시 방출, 필름 코팅, “C2” 각인
- 정제 (250 mg): 흰색, 다이아몬드 모양, 즉시 방출, 필름 코팅, “C25” 각인
- 정제 (300 mg): 노란색, 타원형, 즉시 방출, 필름 코팅, “C3” 각인
4 금기 사항
없음.
5 경고 및 주의 사항
5.1 골수이형성증후군/급성 골수성 백혈병
골수이형성증후군(MDS)/급성 골수성 백혈병(AML)은 Rubraca로 치료받는 환자에게 발생하며, 잠재적으로 치명적인 유해 반응입니다. 난소암으로 치료받은 1594명의 환자에서
[유해 반응(
6.1) 참조]
, MDS/AML은 장기 추적 관찰을 포함하여 32명의 환자(2%)에서 발생했습니다. 이 중 14건은 치료 중 또는 28일 안전성 추적 관찰 기간 동안 발생했습니다(0.9%). MDS/AML 진단 전 Rubraca 치료 기간은 2개월 미만에서 약 72개월까지 다양했습니다. 이러한 사례는 이차 MDS/암 치료 관련 AML의 전형적인 사례였습니다. 모든 사례에서 환자는 이전에 백금 함유 화학 요법 요법 및/또는 기타 DNA 손상제를 투여받았습니다.
ARIEL3에서 생식 세포 및/또는 체세포
BRCA돌연변이가 있는 환자 중 Rubraca로 치료받은 환자의 경우, MDS/AML은 Rubraca로 치료받은 129명 중 9명(7%)에서 발생했고, 위약으로 치료받은 66명 중 4명(6%)에서 발생했습니다. 이차 MDS/암 치료 관련 AML이 발생한 환자의 Rubraca 치료 기간은 1.2년에서 4.7년까지 다양했습니다.
TRITON2에서 MDS/AML은 상동 재조합 결핍(HRD) 돌연변이와 관계없이 mCRPC 환자(n=209)에서 관찰되지 않았습니다.
[유해 반응(
6.1) 참조].
환자가 이전 화학 요법으로 인한 혈액학적 독성(≤ 1등급)에서 회복될 때까지 Rubraca를 시작하지 마십시오. 치료 중 임상적으로 유의미한 변화에 대해 기준선에서 매월 완전 혈액 검사를 모니터링하여 혈구 감소증을 확인하십시오. 장기간 혈액학적 독성(> 4주)의 경우,
표 1[용량 및 투여(
2.3) 참조]
에 따라 Rubraca를 중단하거나 용량을 줄이고 회복될 때까지 매주 혈액 검사를 모니터링하십시오. 4주 후에도 수치가 1등급 이하로 회복되지 않거나 MDS/AML이 의심되는 경우, 골수 분석 및 혈액 샘플의 세포 유전학 검사를 포함한 추가 검사를 위해 환자를 혈액학자에게 의뢰하십시오. MDS/AML이 확인되면 Rubraca를 중단하십시오.
5.2 배아-태아 독성
Rubraca는 작용 기전과 동물 연구 결과를 기반으로 임산부에게 투여하면 태아에게 해를 끼칠 수 있습니다. 동물 생식 연구에서, 기관 형성 기간 동안 임신한 랫트에게 rucaparib를 투여한 결과, 하루 2회 600mg의 권장 인간 용량을 투여받은 환자의 AUC
0-24h의 0.04배에 해당하는 노출량에서 배아-태아 사망이 발생했습니다. 임산부에게 태아에 대한 잠재적 위험을 알리십시오. 임신 가능성이 있는 여성에게는 Rubraca 치료 중 및 마지막 투여 후 6개월 동안 효과적인 피임법을 사용하도록 조언하십시오.
[특정 인구 집단에서의 사용(
8.1,
8.3) 및 임상 약리학(
12.1) 참조].
유전 독성 및 동물 생식 연구 결과를 기반으로, 임신 가능성이 있는 여성 파트너가 있거나 임신한 여성 파트너가 있는 남성 환자에게는 Rubraca 치료 중 및 마지막 투여 후 3개월 동안 효과적인 피임법을 사용하도록 조언하십시오.
[특정 인구 집단에서의 사용(
8.1,
8.3) 참조].
6 부작용
다음의 중대한 이상반응은 라벨의 다른 곳에서 논의됩니다.
- 골수이형성증후군/급성 골수성 백혈병
[경고 및 주의사항 (
5.1) 참조].
6.1 임상시험 경험
임상시험은 매우 다양한 조건에서 수행되므로 임상시험에서 관찰된 약물의 이상반응 발생률을 다른 약물의 임상시험에서 관찰된 발생률과 직접 비교할 수 없으며 실제로 관찰된 발생률을 반영하지 않을 수 있습니다.
경고 및 주의사항 섹션에서 난소암 환자에 대한 통합 안전성 모집단은 ARIEL3을 포함한 임상시험에서 600mg을 1일 2회 경구 투여받은 1594명의 환자에서 루브라카에 대한 노출을 반영합니다. 이 그룹에서 환자의 57%는 6개월 이상 노출되었고 33%는 1년 이상 노출되었습니다.
난소암 환자에 대한 통합 안전성 모집단에서 환자의 ≥ 10%에서 가장 흔한 이상반응은 메스꺼움(68%), 무력증/피로(65%), 빈혈/헤모글로빈 감소(45%), AST/ALT 증가(39%), 구토(36%), 설사(29%), 식욕 감소(27%), 혈소판 감소증/혈소판 수 감소(25%), 미각장애(24%), 호중구 감소증/호중구 수 감소(21%), 혈액 크레아티닌 증가(17%), 호흡곤란(16%), 어지러움(14%), 소화불량(11%), 광과민성 반응(10%) 및 백혈구 감소증/백혈구 수 감소(10%)였습니다.
BRCA 변이의 재발성 난소암의 유지 치료
BRCA 변이의 재발성 상피 난소암, 나팔관암 또는 원발성 복막암 환자에 대한 루브라카 유지 치료의 안전성은 유해한 BRCA 변이가 있는 195명의 환자가 질병 진행 또는 허용할 수 없는 독성이 나타날 때까지 루브라카 600mg을 1일 2회 경구 투여(n=129) 또는 위약(n=66)을 투여받는 무작위(2:1), 이중맹검, 위약 대조 연구인 ARIEL3에서 조사되었습니다. 연구 치료 기간 중앙값은 루브라카를 투여받은 환자의 경우 13.6개월(범위: < 1개월~39개월)이었고 위약을 투여받은 환자의 경우 5.5개월이었습니다.
모든 등급의 이상반응으로 인한 용량 중단은 루브라카를 투여받은 환자의 67%와 위약을 투여받은 환자의 14%에서 발생했습니다. 이상반응으로 인한 용량 감소는 루브라카 환자의 57%와 위약 환자의 6%에서 발생했습니다. 루브라카의 용량 중단 또는 용량 감소로 이어지는 가장 흔한 이상반응은 혈소판 감소증(25%), 빈혈(19%), AST/ALT 증가(16%), 피로/무력증(14%) 및 메스꺼움(10%)이었습니다. 이상반응으로 인한 중단은 루브라카 환자의 15%와 위약 환자의 5%에서 발생했습니다. 루브라카로 치료받은 환자에서 가장 자주 중단으로 이어진 구체적인 이상반응은 혈소판 감소증(4%), 메스꺼움(3%) 및 빈혈(2%)이었습니다.
표 2는 환자의 ≥20%에서 발생하는 이상반응을 설명하고,
표 3는 ARIEL3에서 발생하는 환자의 ≥25%에서 발생하는 실험실적 이상을 설명합니다.
|
aNational Cancer Institute Common Terminology Criteria for Adverse Events(NCI CTCAE 버전 4.03) |
||||
|
b이상반응의 의학적 개념을 반영하는 그룹화된 관련 용어로 구성 |
||||
| 이상반응 | 루브라카 N=129 |
위약 N=66 |
||
| 1~4등급a % |
3~4등급 % |
1~4등급a % |
3~4등급 % |
|
| 위장관 장애 | ||||
| 메스꺼움 | 79 | 2 | 29 | 0 |
| 복통/팽만
b |
48 | 3 | 49 | 2 |
| 변비 | 39 | 4 | 36 | 2 |
| 구토 | 37 | 4 | 14 | 0 |
| 설사 | 34 | 2 | 18 | 0 |
| 구내염
b |
28 | 0.8 | 12 | 0 |
| 전신 장애 및 투여 부위 이상 | ||||
| 피로/무력증 | 74 | 9 | 52 | 5 |
| 피부 및 피하 조직 장애 | ||||
| 발진
b |
45 | 0 | 23 | 0 |
| 신경계 장애 | ||||
| 미각장애 | 33 | 0 | 6 | 0 |
| 두통 | 22 | 0 | 15 | 2 |
| 검사 | ||||
| AST/ALT 상승 | 33 | 16 | 3 | 0 |
| 혈액 및 림프계 장애 | ||||
| 빈혈 | 41 | 26 | 6 | 0 |
| 혈소판 감소증 | 35 | 6 | 3 | 0 |
| 호중구 감소증 | 22 | 8 | 6 | 0 |
| 호흡기, 흉부 및 종격동 장애 | ||||
| 비인두염/상기도 감염
b |
29 | 0 | 20 | 2 |
| 대사 및 영양 장애 | ||||
| 식욕 감소 | 23 | 2 | 14 | 0 |
Rubraca로 치료받은 환자의 < 20%에서 발생하는 이상반응에는 불면증(19%), 호흡곤란(17%), 어지러움(15%), 발열(15%), 소화불량(12%), 말초 부종(12%), 우울증(11%)이 있습니다.
|
aCTCAE Grade 1의 실험실적 수치를 가진 환자는 임상 연구에 참여할 수 있었습니다. |
||||
|
bNCI CTCAE 버전 4.03. |
||||
| 실험실 매개변수a | Rubraca N=129 |
위약 N=66 |
||
| 1-4등급b % |
3-4등급 % |
1-4등급b % |
3-4등급 % |
|
| 화학 | ||||
| 크레아티닌 증가 | 96 | 0 | 89 | 0 |
| ALT 증가 | 67 | 11 | 6 | 0 |
| AST 증가 | 59 | 1 | 6 | 0 |
| 콜레스테롤 증가 | 39 | 3 | 20 | 0 |
| ALP 증가 | 39 | 0 | 3 | 0 |
| 혈액학 | ||||
| 헤모글로빈 감소 | 61 | 18 | 14 | 2 |
| 혈소판 감소 | 47 | 2 | 8 | 0 |
| 백혈구 감소 | 39 | 3 | 23 | 0 |
| 호중구 감소 | 38 | 6 | 18 | 2 |
| 림프구 감소 | 33 | 7 | 14 | 2 |
안드로겐 수용체 표적 치료 및 화학 요법 후 BRCA 변이 mCRPC 치료
1일 2회 Rubraca 600mg의 안전성은 단일군 임상시험(TRITON2)에서 평가되었습니다.
[14.2 임상 연구 참조]
. TRITON2에는 HRD 양성 mCRPC 환자 209명이 등록되었으며, 이 중 115명은
BRCA 변이 mCRPC 환자였습니다.
BRCA 변이 mCRPC 환자의 Rubraca 치료 기간 중앙값은 6.5개월(범위 0.5~26.7)이었습니다.
이상 반응으로 인한 사망은 2명(1.7%)에게서 발생했으며, 각각 급성 호흡곤란 증후군과 폐렴으로 인한 사망이었습니다.
Rubraca를 투여받은 환자의 57%에서 이상 반응으로 인한 용량 중단이 발생했습니다. 환자의 >3%에서 용량 중단이 필요한 이상 반응에는 빈혈, 혈소판 감소증, 무력증/피로, 메스꺼움, 구토, 호중구 감소증, ALT/AST 증가, 크레아티닌 증가, 식욕 감소, 급성 신장 손상, 저인산혈증이 포함되었습니다.
Rubraca를 투여받은 환자의 41%에서 이상 반응으로 인한 용량 감소가 발생했습니다. 환자의 >3%에서 용량 감소가 필요한 이상 반응은 빈혈(14%), 무력증/피로(10%), 혈소판 감소증(7%), 메스꺼움(6%), 식욕 감소(4%), 발진(3%)이었습니다.
Rubraca를 투여받은 환자의 8%에서 이상 반응으로 인한 투여 중단이 발생했습니다. Rubraca 투여 중단으로 이어진 이상 반응(ECG QT 연장, 급성 호흡곤란 증후군, 빈혈, 균형 장애, 심부전, 식욕 감소/피로/체중 감소, 백혈구 감소증/호중구 감소증, ALT/AST 증가, 폐렴)은 1명(<1%)을 초과하는 환자에게서 발생하지 않았습니다.
표 4와
5는 TRITON2에서 BRCA 변이 mCRPC 환자의 이상 반응과 검사 이상을 각각 요약한 것입니다.
| 이상 반응 | Rubraca N = 115 |
|
|---|---|---|
| 1~4등급
a |
3~4등급 (%) |
|
|
aNCI CTCAE 버전 4.03. |
||
|
b혈소판 감소증 포함 |
||
|
c물집, 혈액 물집, 피부염, 접촉성 피부염, 습진, 생식기 발진, 수족 피부 홍반 감각 이상 증후군, 광과민 반응, 건선, 발진, 반점구진성 발진, 가려움성 발진, 피부 박리, 피부 병변, 두드러기 포함 |
||
| 전신 장애 및 투여 부위 이상 | ||
| 무력증/피로 | 62 | 9 |
| 위장관 장애 | ||
| 메스꺼움 | 52 | 3 |
| 변비 | 27 | 1 |
| 구토 | 22 | 1 |
| 설사 | 20 | 0 |
| 혈액 및 림프계 장애 | ||
| 빈혈 | 43 | 25 |
| 혈소판 감소증
b |
25 | 10 |
| 대사 및 영양 장애 | ||
| 식욕 감소 | 28 | 2 |
| 피부 및 피하 조직 장애 | ||
| 발진
c |
27 | 2 |
| 검사 | ||
| ALT/AST 증가 | 33 | 5 |
호흡곤란, 어지러움, 출혈, 요로 감염, 미각장애, 소화불량, 과민증(홍조, 천식, 질식감, 눈 주위 부기, 얼굴 부기, 쌕쌕거림 포함), 폐렴, 패혈증, 허혈성 심혈관계 사건, 신부전, 정맥 혈전색전증, 구내염을 포함한 기타 임상적으로 관련된 이상반응이 환자의 20% 미만에서 발생했습니다.
|
a각 검사 항목에 대한 분모는 111~115명의 환자에 대해 사용 가능한 기준치 및 치료 후 검사 값을 기준으로 합니다. |
||
|
bNCI CTCAE 버전 5.0; 인산염 감소는 NCI CTCAE 버전 4.03을 사용하여 등급이 매겨집니다. |
||
|
c3-4등급 ALT 또는 AST 상승으로 인해 4명의 환자에서 약물 투여가 중단되었으며, 이 중 1명은 재투여 시 용량이 감소했습니다. |
||
| 검사 항목 | Rubraca N = 115 a |
|
| 1-4등급
b
|
3-4등급 (%) |
|
| 임상 화학 | ||
| ALT 증가
c |
69 | 5 |
| 인산염 감소 | 68 | 15 |
| 알칼리성 인산분해효소 증가 | 44 | 2 |
| 크레아티닌 증가 | 43 | 2 |
| 트리글리세리드 증가 | 42 | 5 |
| 나트륨 감소 | 38 | 3 |
| 혈액학 | ||
| 백혈구 감소 | 69 | 5 |
| 절대 호중구 수 감소 | 62 | 10 |
| 헤모글로빈 감소 | 59 | 25 |
| 림프구 감소 | 42 | 17 |
| 혈소판 감소 | 40 | 10 |
7 약물 상호 작용
7.1 Rubraca의 다른 약물에 대한 영향
특정 CYP1A2, CYP3A, CYP2C9 또는 CYP2C19 기질
Rubraca와 CYP1A2, CYP3A, CYP2C9 또는 CYP2C19 기질을 동시에 투여하면 이러한 기질의 전신 노출이 증가할 수 있습니다.
[임상 약리학 (
12.3)]
, 이는 이러한 기질의 부작용 빈도 또는 심각도를 증가시킬 수 있습니다. Rubraca와 이러한 효소의 기질을 동시에 투여해야 하지만 최소 농도 변화가 심각한 부작용으로 이어질 수 있는 경우, 승인된 처방 정보에 따라 기질 용량을 줄이십시오.
와파린(CYP2C9 기질)과의 동시 투여를 피할 수 없는 경우, 국제 표준화 비율(INR) 모니터링 빈도를 높이는 것을 고려하십시오.
8. 특정 환자군에서의 사용
8.1 임신
위험 요약
동물 연구 결과 및 작용 기전에 따르면, Rubraca는 임산부에게 투여 시 태아에게 해를 끼칠 수 있습니다. 임산부에서 약물 관련 위험을 알려주는 데이터는 없습니다. 동물 생식 연구에서 임신한 쥐에게 기관 형성 기간 동안 rucaparib을 투여한 결과, 1일 2회 600mg의 권장 용량을 투여받은 환자에서 AUC
0-24h의 0.04배에 해당하는 모체 노출에서 배아-태아 사망이 발생했습니다
[데이터 참조]. 임산부에게 태아에 대한 잠재적 위험을 알립니다.
해당 환자군에 대한 주요 선천적 기형 및 유산의 배경 위험은 알려져 있지 않습니다. 미국 전체 인구에서 임상적으로 인지된 임신의 주요 선천적 기형 및 유산의 추정 배경 위험은 각각 2%~4% 및 15%~20%입니다.
8.5 노인에서의 사용
ARIEL3을 포함한 임상 시험에서 Rubraca를 투여받은 난소암 환자 937명 중 41%가 65세 이상이었고 10%가 75세 이상이었습니다. 젊은 난소암 환자와 노년층 난소암 환자 간에 안전성에서 주요 차이점은 관찰되지 않았습니다.
TRITON2에서 Rubraca를 투여받은 mCRPC 환자 209명 중 77%가 65세 이상이었고 33%가 75세 이상이었습니다. 젊은 mCRPC 환자와 노년층 mCRPC 환자 간에 안전성에서 주요 차이점은 관찰되지 않았습니다.
8.6 신장애
경증에서 중등도의 신장애(Cockcroft-Gault 방법으로 추정한 크레아티닌 청소율 [CLcr] 30~89mL/min) 환자의 경우 용량 조절이 권장되지 않습니다
[임상 약리학(
12.3) 참조]
. CLcr < 30mL/min인 환자 또는 투석을 받는 환자에서는 Rubraca가 연구되지 않았습니다.
8.7 간장애
경증에서 중등도의 간장애(총 빌리루빈 ≤ 정상 상한치[ULN]의 3배 또는 AST > ULN) 환자의 경우 용량 조절이 권장되지 않습니다
[임상 약리학(
12.3) 참조]
. 중증 간장애(총 빌리루빈 > ULN의 3배 및 AST) 환자에서는 Rubraca가 연구되지 않았습니다.
11 설명
루카파립은 포유류 폴리아데노신 5′-디포스포리보실폴리머라제(PARP) 효소의 억제제입니다. 화학 이름은 8-플루오로-2-{4-[(메틸아미노)메틸]페닐}-1,3,4,5-테트라히드로-6H-아제피노[5,4,3-cd]인돌-6-온((1S,4R)-7,7-디메틸-2-옥소바이사이클로[2.2.1]헵트-1-일)메탄설폰산염입니다. 루카파립 캄실레이트의 화학식은 C
19H
18FN
3O•C
10H
16O
4S이며 상대 분자량은 555.67 달톤입니다.
루카파립 캄실레이트의 화학 구조는 아래와 같습니다.
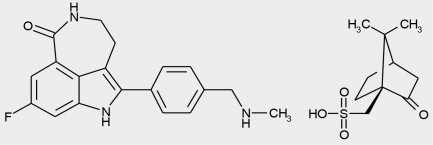
루카파립 캄실레이트는 흰색에서 연한 노란색 분말이며, 경구 투여를 위한 정제로 제형화됩니다. 루카파립은 생리적 pH 범위에서 약 1 mg/mL의 pH 의존성이 낮은 용해도를 나타냅니다.
루브라카(루카파립) 정제는 활성 성분으로 루카파립 캄실레이트를 함유하고 있습니다. 200 mg 정제는 200 mg 루카파립 유리 염기와 동일한 344 mg 루카파립 캄실레이트를 함유하고 있습니다. 250 mg 정제는 250 mg 루카파립 유리 염기와 동일한 430 mg 루카파립 캄실레이트를 함유하고 있습니다. 300 mg 정제는 300 mg 루카파립 유리 염기와 동일한 516 mg 루카파립 캄실레이트를 함유하고 있습니다.
루브라카 정제의 비활성 성분은 다음과 같습니다: 미결정 셀룰로오스, 나트륨 전분 글리콜레이트, 콜로이드성 실리카, 마그네슘 스테아레이트. 200 mg 정제의 화장품 블루 필름 코팅, 250 mg 정제의 화장품 화이트 필름 코팅, 300 mg 정제의 화장품 옐로우 필름 코팅은 폴리비닐 알코올, 이산화티타늄, 폴리에틸렌 글리콜/마크로골, 활석을 함유한 Opadry II입니다. 코팅은 푸른색 밝은 파란색 알루미늄 호수와 인디고 카민 알루미늄 호수를 사용하여 푸른색으로 착색되거나, 노란색 산화철을 사용하여 노란색으로 착색됩니다.
12. 임상 약리학
12.1 작용 기전
루카파립은 DNA 복구에 역할을 하는 PARP-1, PARP-2 및 PARP-3을 포함한 폴리(ADP-리보스) 폴리머라제(PARP) 효소의 억제제입니다.
시험관 내연구에 따르면 루카파립 유도 세포독성은 PARP 효소 활성 억제 및 PARP-DNA 복합체 형성 증가를 포함할 수 있으며, 이는 DNA 손상, 세포자멸사 및 암세포 사멸로 이어집니다.
BRCA1/2및 기타 DNA 복구 유전자 결핍이 있는 종양 세포주에서 루카파립 유도 세포독성 및 항종양 활성이 증가했습니다. 루카파립은
BRCA결핍 여부에 관계없이 인간 암의 마우스 이종 이식 모델에서 종양 성장을 감소시키는 것으로 나타났습니다.
12.3 약동학
루카파립의 AUC 및 C
max는 하루에 두 번 240mg에서 840mg(승인된 권장 용량의 0.4배에서 1.4배)까지의 용량 범위에서 선형 약동학을 보였습니다. 승인된 권장 용량에서 평균(변동 계수 [CV]) 안정 상태 루카파립 C
max는 1,940ng/mL(54%)이고 AUC
0-12h는 16,900h×ng/mL(54%)입니다. 평균 AUC 누적 비율은 3.5배에서 6.2배입니다.
흡수
승인된 권장 용량에서 안정 상태에서 중앙값 T
max(최소, 최대)는 1.9시간(0, 5.98)입니다. 평균(최소, 최대) 절대 생체 이용률은 36%(30%, 45%)입니다.
음식의 영향
고지방 식사(약 800-1000칼로리, 탄수화물에서 약 250칼로리, 지방에서 약 500-600칼로리, 단백질에서 약 150칼로리 포함) 후 C
max는 20% 증가했고 AUC
0-24h는 38% 증가했으며 T
max는 금식 상태에 비해 2.5시간 지연되었습니다.
[용법 및 용량(
2.2) 참조]
.
분포
평균 명목상 분포 용적은 2300L(21%)입니다.
루카파립은 시험관 내에서 인간 혈장 단백질에 70% 결합합니다. 루카파립은 혈액-혈장 농도 비율이 1.8인 적혈구에 우선적으로 분포합니다.
대사
시험관 내에서 루카파립은 주로 CYP2D6에 의해 대사되고 CYP1A2 및 CYP3A4에 의해 소량 대사됩니다. CYP 기반 산화 외에도 루카파립은 N-탈메틸화, N-메틸화 및 글루쿠론화를 거칩니다.
배출
방사성 표지된 루카파립을 단일 경구 투여한 후, 변하지 않은 루카파립은 방사성 동위원소의 64%를 차지했습니다. 루카파립은 각각 소변과 대변에서 방사성 동위원소의 45%와 95%를 차지했습니다.
특정 인구 집단
연령(20세에서 86세), 인종(백인, 흑인 및 아시아인), 성별, 체중(41kg에서 171kg), 경도에서 중등도 신장애(CLcr ≥ 30mL/min), 경도 간 기능 장애(총 빌리루빈 < ULN 및 AST > ULN 또는 총 빌리루빈 1에서 1.5 x ULN 및 모든 AST), CYP2D6 또는 CYP1A2 유전자형 다형성은 루카파립의 약동학에 임상적으로 의미 있는 영향을 미치지 않았습니다. 중증 신장애(CLcr 15에서 29mL/min), 말기 신장 질환(CLcr < 15mL/min) 또는 중증 간 기능 장애(총 빌리루빈 > 3 x ULN 및 모든 AST)의 영향은 연구되지 않았습니다.
간 기능 장애
중등도 간 기능 장애(총 빌리루빈 > 1.5에서 3 x ULN 및 모든 AST)는 루카파립 AUC를 45% 증가시켰지만 정상 간 기능이 있는 환자에 비해 C
max에는 영향을 미치지 않았습니다.
루카파립의 다른 약물에 대한 영향
루브라카와 로수바스타틴(BCRP 기질)을 병용 투여하면 로수바스타틴의 농도에 임상적으로 의미 있는 영향을 미치지 않았습니다.
루카파립과 다음 기질을 함께 투여하면 각 공동 투여된 기질의 C
max가 ≤ 1.1배 증가하고 각 기질의 AUC가 다음과 같이 증가했습니다.
- 카페인 (CYP1A2): 2.6배 증가
- 미다졸람 (CYP3A4): 1.4배 증가
- 와파린 (CYP2C9): 1.5배 증가
- 오메프라졸 (CYP2C19): 1.6배 증가
- 디곡신 (P-glycoprotein): 1.2배 증가
루카파립과 에티닐에스트라디올과 레보노르게스트렐(CYP3A 기질)을 함유한 경구 피임약을 함께 투여하면 에티닐에스트라디올 AUC가 1.4배, 레보노르게스트렐 AUC가 1.6배 증가했지만, 임상적으로 의미 있는 C
max에 대한 영향은 없었습니다.
시험관 내 연구
시토크롬 P450 (CYP) 효소: 루카파립은 CYP2C8 및 CYP2D6을 억제하고 CYP1A2를 유도했습니다.
UDP-글루쿠로노실전이효소 (UGT) 효소: 루카파립은 UGT1A1을 억제했습니다.
수송체 시스템: 루카파립은 P-gp 및 BCRP의 기질입니다. 루카파립은 OATP1B1, OATP1B3, OAT1, OAT3 또는 OCT2의 기질이 아닙니다.
루카파립은 OATP1B1, OATP1B3, OAT1, OAT3, MATE1, MATE2-K, OCT1, OCT2 및 MRP4를 억제했습니다. 루카파립은 MRP2, MRP3 또는 BSEP을 억제하지 않았습니다.
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 저해
루카파립으로 발암성 연구는 수행되지 않았습니다.
루카파립은 배양된 인간 림프구에서
in vitro염색체 이상 분석에서 클라스토젠성을 나타냈습니다. 유사분열 자극 세포에서의 클라스토젠성 반응은 루카파립의 작용 기전을 기반으로 예상되었으며, 인간에게 잠재적인 유전 독성을 나타냅니다. 루카파립은 박테리아 역 돌연변이(Ames) 시험에서 돌연변이 유발성이 없었습니다.
루카파립으로 생식능력 연구는 수행되지 않았습니다. 3개월 반복 투여 일반 독성 연구에서 루카파립은 각각 랫드와 개에서 100 mg/kg/day 및 20 mg/kg/day까지의 용량에서 수컷 및 암컷 생식 기관에 영향을 미치지 않았습니다. 이러한 용량 수준은 권장 용량에서 각각 약 0.3배 및 0.09배의 인체 노출(AUC
0-24h)에 해당합니다.
14 임상 연구
14.1 BRCA 돌연변이 재발성 난소암의 유지 치료
Rubraca의 효능은 ARIEL3 (NCT01968213)에서 조사되었으며, 이는 백금 기반 화학 요법에 반응한 재발성 상피성 난소암, 난관암 또는 원발성 복막암 환자 564명을 대상으로 한 이중맹검, 다기관 임상 시험으로, 환자들은 Rubraca 정제 600mg을 1일 2회 경구 투여 (n=375) 또는 위약 (n=189)을 받도록 무작위 배정 (2:1)되었습니다. 치료는 질병 진행 또는 용납할 수 없는 독성이 나타날 때까지 계속되었습니다. 모든 환자는 가장 최근 백금 기반 화학 요법에 반응 (완전 또는 부분)을 보였습니다. 무작위 배정은 마지막 백금에 대한 최상의 반응 (완전 또는 부분), 마지막 백금 치료 후 진행까지의 시간 (6개월에서 ≤ 12개월 및 > 12개월), 종양 바이오 마커 상태에 따라 계층화되었습니다.
종양 조직 샘플은 임상 시험 분석 (CTA) (N=564) 및 연구용 Foundation Medicine 조직 검사 (n=518)를 사용하여 검사했습니다. 두 검사 모두로 평가된 샘플 중 종양
BRCA(t
BRCA) 돌연변이 상태는 CTA로 결정된 t
BRCA 양성 환자의 99% (177/178)에서 확인되었습니다. t
BRCA 환자의 94% (186/196)에 대한 혈액 샘플은 중앙 혈액 배선
BRCA 검사를 사용하여 평가했습니다. 이러한 결과를 바탕으로 t
BRCA 환자의 70% (130/186)는 배선
BRCA 돌연변이가 있었고 30% (56/186)는 체세포
BRCA 돌연변이가 있었습니다. 효능 결과는 t
BRCA(배선 또는 체세포) 하위 그룹을 기반으로 합니다.
주요 효능 결과는 고형 종양에 대한 반응 평가 기준 (RECIST), 버전 1.1 (v1.1)에 따라 평가된 연구자 평가 진행 무료 생존율 (PFS)이었습니다. 전체 생존율 (OS)은 추가 결과 측정 항목이었습니다.
등록된 564명의 환자 중 196명 (35%)은 t
BRCA 돌연변이가 있었습니다.
tBRCA 돌연변이가 있는 환자 중 Rubraca를 투여받은 환자의 중간 연령은 58세 (범위: 42세에서 81세)였고 위약을 투여받은 환자의 중간 연령은 59세 (범위: 36세에서 84세)였습니다. 대부분이 백인 (84%)이었고 100%가 ECOG 수행 상태가 0 또는 1이었습니다. 모든 환자는 이전에 백금 기반 화학 요법을 2회 이상 (범위: 2회에서 5회) 받았습니다. 환자의 33%가 가장 최근 치료에 완전 반응 (CR)을 보였습니다. 마지막 백금까지의 진행 무료 간격은 환자의 41%에서 6-12개월이었고 59%에서 > 12개월이었습니다. Rubraca를 투여받은 환자의 22%와 위약을 투여받은 환자의 17%에서 이전 베바시주맙 치료가 보고되었습니다. 기준선에서 측정 가능한 질병이 환자의 32%에서 나타났습니다.
ARIEL3는 t
BRCA 돌연변이가 있는 환자에서 Rubraca를 무작위 배정받은 환자와 위약을 무작위 배정받은 환자를 비교했을 때 PFS가 통계적으로 유의미하게 개선되었음을 보여주었습니다. 맹검된 독립적인 방사선학적 검토 결과가 일치했습니다.
t
BRCA 돌연변이가 있는 환자에 대한 효능 결과는
표 6 및
그림 1에 요약되어 있습니다.
|
at BRCA는 CTA로 결정된 유해한 배선 또는 체세포 BRCA 돌연변이가 있는 모든 환자를 포함합니다. |
||
| Rubraca N=130 |
위약 N=66 |
|
| 진행 무료 생존율 | ||
| 이벤트 수, n (%) | 67 (52%) | 56 (85%) |
| 중간값 (월) (95% CI) | 16.6 (13.4-22.9) | 5.4 (3.4-6.7) |
| HR (95% CI) | 0.23 (0.16, 0.34) | |
| p-값 | < 0.0001 | |
Figure 1. Kaplan-Meier Curves of Progression-Free Survival in ARIEL3 as Assessed by Investigator: tBRCAGroup
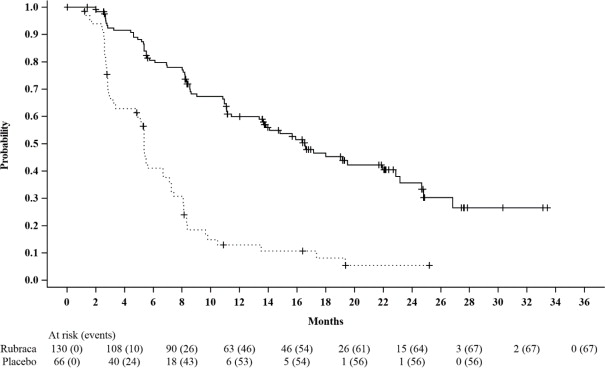
A final OS analysis was conducted after 130 events were observed. Exploratory OS results showed a HR of 0.83 (95% CI: 0.58, 1.19) in the t
BRCAsubgroup with a median OS of 45.9 months (95% CI: 37.7, 59.6) for patients treated with Rubraca and 47.8 months (95% CI: 43.2, 55.8) for patients on placebo.
14.2
BRCA-mutated Metastatic Castration-Resistant Prostate Cancer
The efficacy of Rubraca was investigated in TRITON2 (NCT02952534), an ongoing multi-center, single arm clinical trial in patients with
BRCA-mutated mCRPC who had been treated with androgen receptor-directed therapy and taxane-based chemotherapy. There were 115 patients with either germline or somatic
BRCAmutations enrolled in TRITON2, of whom 62 patients had measurable disease at baseline by independent radiology review (IRR). Patients received Rubraca 600 mg orally twice daily until disease progression or unacceptable toxicity. Patients also received concomitant GnRH analog or had prior bilateral orchiectomy. Objective response rate (ORR) and duration of response (DOR) were assessed in patients with measurable disease by blinded IRR and by the investigator according to modified RECIST v1.1/ Prostate Cancer Working Group 3 (PCWG3) criteria.
For the 62 patients with measurable disease at baseline, the median age was 73 years (range 52 to 88); the majority were White (73%) and 10% were Black; and 98% of patients had an ECOG performance status of 0 or 1. All patients had received at least one prior androgen receptor-directed therapy, 34% had received 2 prior androgen receptor-directed therapies and 2% had received 3 prior androgen receptor-directed therapies, and all patients also received prior taxane chemotherapy. Eighteen percent of patients had lung and 21% had liver metastases at baseline. Twenty-four percent had metastases to lymph nodes alone. Forty percent had 10 or more bone lesions at baseline.
All 62 patients had a deleterious somatic or germline
BRCAmutation detected from either central plasma (26%), central tissue (32%), or local (42%) testing. Of the 62 patients, 66% had a somatic
BRCAmutation, 34% had a germline
BRCAmutation, 85% had a
BRCA2mutation, and 15% had a
BRCA1mutation.
The major efficacy outcomes of the study were confirmed ORR by IRR using modified RECIST v1.1/PCWG3 criteria and DOR. Efficacy results of TRITON2 are provided in
Table 7. The ORR by IRR was similar in patients with germline versus somatic
BRCAmutation.
|
NE = not evaluable |
|
|
aDefined per modified RECIST v1.1 criteria and with no confirmed bone progression per PCWG3. |
|
|
bThe range for the DOR was 1.7-24+ months. Fifteen of the 27 (56%) patients with a confirmed objective response had a DOR of ≥ 6 months. |
|
| Rubraca (N = 62) |
|
| Confirmed Objective Response Rate (95% CI)
a |
44% (31, 57) |
| Median DOR in months (95% CI)
b |
NE (6.4, NE) |
16. 공급/보관 및 취급 방법
공급 형태
Rubraca는 200mg, 250mg 및 300mg 정제로 제공됩니다.
200mg 정제:
- 한쪽 면에 “C2”가 각인된 파란색 원형 정제
- 60정들이 병에 포장됨 (NDC: 69660-201-91)
250mg 정제:
- 한쪽 면에 “C25”가 각인된 흰색 다이아몬드형 정제
- 60정들이 병에 포장됨 (NDC: 69660-202-91)
300mg 정제:
- 한쪽 면에 “C3”가 각인된 노란색 타원형 정제
- 60정들이 병에 포장됨 (NDC: 69660-203-91)
17 환자 상담 정보
환자에게 FDA 승인 환자 라벨(
환자 정보)을 읽도록 알려주십시오.
MDS/AML:환자가 쇠약감, 피로감, 발열, 체중 감소, 잦은 감염, 멍, 쉽게 출혈, 숨가쁨, 소변이나 대변에 피가 섞여 나오는 증상, 또는 낮은 혈구 수치의 실험실 검사 결과 또는 수혈이 필요한 경우 의료 서비스 제공자에게 연락하도록 알려주십시오. 이는 혈액학적 독성 또는 Rubraca로 치료받은 환자에게 보고된 ‘골수 이형성 증후군'(MDS) 또는 ‘급성 골수성 백혈병'(AML)이라는 더 심각한 드문 골수 문제의 징후일 수 있습니다.
[경고 및 주의 사항(
5.1) 참조]
.
태아-태아 독성:여성에게 임신 중이거나 임신하게 되면 의료 서비스 제공자에게 알리도록 알려주십시오. 여성 환자에게 태아에 대한 위험과 임신 손실 가능성을 알려주십시오.
[특정 인구 집단에서의 사용(
8.1) 참조]
. 생식 가능성이 있는 여성에게는 Rubraca 치료 중 및 마지막 Rubraca 투여 후 6개월 동안 효과적인 피임법을 사용하도록 알려주십시오. 생식 가능성이 있는 여성 파트너가 있거나 임신한 여성 파트너가 있는 남성 환자에게는 Rubraca 치료 중 및 마지막 Rubraca 투여 후 3개월 동안 효과적인 피임법을 사용하도록 알려주십시오. 남성 환자에게는 치료 중 및 마지막 Rubraca 투여 후 3개월 동안 정자를 기증하지 않도록 알려주십시오.
[경고 및 주의 사항(
5.2) 및 특정 인구 집단에서의 사용(
8.1,
8.3) 참조]
.
투약 지침:환자에게 Rubraca를 식사와 관계없이 하루 두 번 경구로 복용하도록 지시하십시오. 투약은 약 12시간 간격으로 이루어져야 합니다. 환자에게 Rubraca 복용량을 놓치거나 Rubraca 복용 후 구토하는 경우 추가 복용량을 복용하지 말고 다음 복용량을 정해진 시간에 복용하도록 알려주십시오.
[투약 및 투여(
2.2) 참조]
.
배포:
Clovis Oncology, Inc.
Boulder, CO 80301
1-844-258-7662
Rubraca는 Clovis Oncology, Inc.의 등록 상표입니다.
환자용 요약 정보
|
본 환자 정보는 미국 식품의약국(FDA)의 승인을 받았습니다. |
개정: 2022년 12월 |
|
| 환자 정보 Rubraca®(루브라카) (루카파립) 정제 |
||
|
Rubraca에 대해 알아야 할 가장 중요한 정보는 무엇입니까? Rubraca는 다음과 같은 심각한 부작용을 유발할 수 있습니다. |
||
|
|
|
의료진은 혈구 수치를 확인하기 위해 혈액 검사를 실시합니다.
참조 “Rubraca의 가능한 부작용은 무엇입니까?”부작용에 대한 자세한 내용은. |
||
| Rubraca는 무엇입니까? Rubraca는 성인에게 사용되는 처방약으로 다음과 같은 경우에 사용됩니다.
Rubraca가 어린이에게 안전하고 효과적인지 여부는 알려져 있지 않습니다. |
||
Rubraca를 복용하기 전에 의료진에게 모든 의학적 상태를 알리십시오. 다음과 같은 경우 포함:
복용하는 모든 약물을 의료진에게 알리십시오.처방약과 일반 의약품, 비타민 및 허브 보충제를 포함합니다. |
||
Rubraca는 어떻게 복용해야 합니까?
|
||
| 루브라카 복용 시 주의 사항은 무엇입니까? 햇볕에 오래 노출되는 것을 피하십시오. 루브라카는 피부를 햇볕에 민감하게 만들 수 있습니다(광과민성). 루브라카 치료 중에는 햇볕에 더 쉽게 탈 수 있습니다. 햇볕에 나가야 할 경우 모자와 피부를 가리는 옷을 착용하고 자외선 차단제를 사용하여 햇볕으로부터 피부를 보호하십시오. |
||
|
루브라카는 심각한 부작용을 유발할 수 있습니다. 난소암 환자에서 루브라카의 가장 흔한 부작용은 다음과 같습니다. |
||
|
|
|
| 전립선암 환자에서 루브라카의 가장 흔한 부작용은 다음과 같습니다. |
||
|
|
|
| 이것들은 루브라카의 모든 가능한 부작용이 아닙니다. 자세한 내용은 의료 서비스 제공자 또는 약사에게 문의하십시오. 부작용에 대한 의학적 조언은 의료 서비스 제공자에게 문의하십시오. FDA에 부작용을 보고할 수 있습니다. 전화번호는 1-800-FDA-1088입니다. |
||
루브라카는 어떻게 보관해야 합니까?
루브라카와 모든 의약품은 어린이의 손이 닿지 않는 곳에 보관하십시오. |
||
| 루브라카의 안전하고 효과적인 사용에 대한 일반 정보. 의약품은 때때로 환자 정보 팜플렛에 나열된 목적 이외의 목적으로 처방됩니다. 루브라카는 처방된 목적 이외의 질환에 사용하지 마십시오. 동일한 증상이 있어도 다른 사람에게 루브라카를 주지 마십시오. 해당 사람에게 해로울 수 있습니다. 루브라카에 대한 자세한 내용은 의료 서비스 제공자 또는 약사에게 문의하십시오. |
||
| 루브라카의 성분은 무엇입니까? 활성 성분:루카파립 비활성 성분:미결정 셀룰로오스, 나트륨 전분 글리콜레이트, 콜로이드성 실리카 및 마그네슘 스테아레이트. 필름 코팅에는 폴리비닐 알코올, 이산화티탄, 폴리에틸렌 글리콜/마크로골 및 활석이 포함됩니다. 파란색 필름 코팅에는 밝은 파란색 알루미늄 호수와 인디고 카민 알루미늄 호수가 포함됩니다. 노란색 필름 코팅에는 황색 산화철이 포함됩니다. 배포: Clovis Oncology, Inc. Boulder, Colorado 80301 자세한 내용은 www.Rubraca.com을 방문하거나 1-844-258-7662로 전화하십시오. |
||
주요 표시면
주요 표시 판넬 – Rubraca 정 200mg 병 라벨
NDC 69660-
201-91
Rubraca®
(루카파리브)정제
200 mg*
60 정제
각 정제는 344 mg 루카파리브 캄실레이트를 함유하며 200 mg 루카파리브와 동등합니다.
의사 처방전에 의해서만
어린이의 손이 닿지 않는 곳에 보관

주요 표시면
주요 표시판 – Rubraca 250mg 정제병 라벨
NDC 69660-
202-91
Rubraca®
(루카파리브)정제
250mg*
60정
각 정제에는 430mg의 루카파리브 캄실레이트가 250mg의 루카파리브와 동등합니다.
전문의处방전용
어린이의 손이 닿지 않는 곳에 보관

주요 표시면
주요 디스플레이 패널 – 루브라카 정제 300 mg 병 라벨
NDC 69660-
203-91
루브라카®
(루카파립)정제
300 mg*
60 정제
각 정제에는 300 mg 루카파립에 해당하는 516 mg 루카파립 카멜실레이트 함유
처방전 전용
어린이 손이 닿지 않는 곳에 보관하세요
