의약품 제조업체: AstraZeneca Pharmaceuticals LP (Updated: 2023-11-06)
처방 정보 하이라이트
린파자®(올라파립) 정제, 경구 복용
최초 미국 승인: 2014년
최근 주요 변경 사항
적응증 및 사용법
린파자는 폴리(ADP-리보오스) 폴리머라제(PARP) 억제제로 다음과 같은 적응증이 있습니다.
난소암
- •
- 첫 번째 백금 기반 화학요법에 완전 또는 부분 반응을 보인 삼중음성 또는 의심되는 삼중음성 생식계 또는 체세포 BRCA 변이가 있는 진행성 상피성 난소암, 나팔관암 또는 원발성 복막암 성인 환자의 유지 요법. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.1, 2.1)
- •
- 베바시주맙과 병용하여 첫 번째 백금 기반 화학요법에 완전 또는 부분 반응을 보인 진행성 상피성 난소암, 나팔관암 또는 원발성 복막암 성인 환자의 유지 요법으로 사용하며, 이들 암은 다음 중 하나로 정의된 상동재조합결함(HRD) 양성 상태와 관련이 있습니다:
- •
- 삼중음성 또는 의심되는 삼중음성 BRCA 변이, 및/또는
- •
- 유전체 불안정성.
- 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.2, 2.1)
- •
- 백금 기반 화학요법에 완전 또는 부분 반응을 보인 삼중음성 또는 의심되는 삼중음성 생식계 또는 체세포 BRCA 변이가 있는 재발성 상피성 난소암, 나팔관암 또는 원발성 복막암 성인 환자의 유지 요법. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.3, 2.1)
유방암
- •
- 선행 또는 보조 화학요법을 받은 삼중음성 또는 의심되는 삼중음성 BRCAm 인간 상피성장인자 수용체 2(HER2) 음성 고위험 초기 유방암 성인 환자의 보조 치료. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.4, 2.1)
- •
- 선행 또는 보조 화학요법 또는 전이 치료 과정에서 치료를 받은 삼중음성 또는 의심되는 삼중음성 gBRCAm HER2 음성 전이성 유방암 성인 환자의 치료. 호르몬 수용체(HR) 양성 유방암 환자는 이전에 내분비 요법을 받았거나 내분비 요법이 적합하지 않은 경우여야 합니다. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.5, 2.1)
췌장암
- •
- 첫 번째 백금 기반 화학요법 요법으로 최소 16주 동안 질병이 진행되지 않은 삼중음성 또는 의심되는 삼중음성 gBRCAm 전이성 췌장선암 성인 환자의 유지 요법. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.6, 2.1)
전립선암
- •
- 이전 엔자루타마이드 또는 아비라테론 치료 후 진행된 삼중음성 또는 의심되는 삼중음성 생식계 또는 체세포 상동재조합 복구(HRR) 유전자 변이가 있는 전이성 거세 내성 전립선암(mCRPC) 성인 환자의 치료. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.7, 2.1)
- •
- 아비라테론 및 프레드니손 또는 프레드니솔론과 병용하여 삼중음성 또는 의심되는 삼중음성 BRCA 변이(BRCA m)가 있는 전이성 거세 내성 전립선암(mCRPC) 성인 환자의 치료. 린파자 치료 대상 환자를 선별하기 위해 FDA 승인 동반진단을 사용하십시오. (1.8, 2.1)
투여량 및 투여 방법
제형 및 농도
정제: 150mg, 100mg (3)
금기 사항
없음. (4)
경고 및 주의사항
- •
- 골수이형성증후군/급성골수성백혈병(MDS/AML): 린파자에 노출된 각종 BRCAm, gBRCAm, HRR 유전자 변이 또는 HRD-양성 암 환자의 약 1.2%에서 발생했으며 대부분의 사례가 치명적이었습니다. 기저 시점과 그 후 매월 혈액학적 독성을 모니터링하십시오. MDS/AML이 확인되면 투여를 중단하십시오. (5.1)
- •
- 폐렴: 린파자에 노출된 환자의 0.8%에서 발생했으며 일부 사례는 치명적이었습니다. 폐렴이 의심되면 치료를 중단하십시오. 폐렴이 확인되면 투여를 중단하십시오. (5.2)
- •
- 중증 또는 치명적인 폐색전증을 포함한 정맥혈전색전증(VTE)이 린파자 치료 환자에서 발생했습니다. mCRPC 환자의 8%에서 VTE가 발생했습니다. 환자의 VTE 및 PE 징후와 증상을 모니터링하고 적절히 치료하십시오. (5.3)
- •
- 태아 독성: 태아에게 해를 끼칠 수 있습니다. 태아에 대한 잠재적 위험과 효과적인 피임법 사용에 대해 알리십시오. (5.4, 8.1, 8.3)
이상반응
임상시험에서 가장 흔한 이상반응(≥10%)은:
- •
- 단일요법으로는 구역, 피로(무력증 포함), 빈혈, 구토, 설사, 식욕감퇴, 두통, 미각장애, 기침, 호중구감소증, 호흡곤란, 현기증, 소화불량, 백혈구감소증, 혈소판감소증이었습니다. (6.1)
- •
- 베바시주맙 병용요법에서는 구역, 피로(무력증 포함), 빈혈, 림프구감소증, 구토, 설사, 호중구감소증, 백혈구감소증, 요로감염, 두통이었습니다. (6.1)
- •
- 아비라테론 및 프레드니손 또는 프레드니솔론 병용요법에서는 빈혈, 피로, 구역, 설사, 식욕감퇴, 림프구감소증, 현기증, 복통이었습니다. (6.1)
의심되는 이상반응은 아스트라제네카 1-800-236-9933 또는 FDA 1-800-FDA-1088 또는 www.fda.gov/medwatch로 보고하십시오.
약물상호작용
환자 상담 정보 및 의약품 가이드는 17항을 참조하십시오.
개정: 2023년 11월
목차
전체 처방 정보: 목차*
1 적응증 및 용법
1.1 BRCA 돌연변이 진행성 난소암의 일차 유지요법
1.2 Bevacizumab과 병용한 HRD 양성 진행성 난소암의 일차 유지요법
1.3 BRCA 돌연변이 재발성 난소암의 유지요법
1.4 생식세포 BRCA 돌연변이 HER2 음성 고위험 초기 유방암의 수술 후 보조요법
1.5 생식세포 BRCA 돌연변이 HER2 음성 전이성 유방암
1.6 생식세포 BRCA 돌연변이 전이성 췌장 선암종의 일차 유지요법
1.7 HRR 유전자 돌연변이 전이성 거세저항성 전립선암
1.8 Abiraterone 및 Prednisone 또는 Prednisolone과 병용한 BRCA 돌연변이 전이성 거세저항성 전립선암 치료
2 용법 및 용량
2.1 환자 선택
2.2 권장 용량
2.3 이상반응에 대한 용량 조절
2.4 강력한 또는 중등도 CYP3A 억제제와 병용 시 용량 조절
2.5 신장애에 대한 용량 조절
3 제형 및 함량
4 금기사항
5 경고 및 주의사항
5.1 골수형성이상증후군/급성 골수성 백혈병
5.2 폐렴
5.3 정맥 혈전색전증
5.4 태아 독성
6 이상반응
6.1 임상시험 경험
6.2 시판 후 경험
7 약물 상호작용
7.1 항암제와의 병용
7.2 Lynparza에 대한 다른 약물의 영향
8 특정 집단에서의 사용
8.1 임신
8.2 수유
8.3 가임기 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 신장애
8.7 간장애
11 설명
12 임상 약리학
12.1 작용기전
12.2 약력학
12.3 약동학
13 비임상 독성학
13.1 발암성, 변이원성, 수태능 장애
14 임상 연구
14.1 BRCA 돌연변이 진행성 난소암의 일차 유지요법
14.2 Bevacizumab과 병용한 HRD 양성 진행성 난소암의 일차 유지요법
14.3 BRCA 돌연변이 재발성 난소암의 유지요법
14.4 생식세포 BRCA 돌연변이 HER2 음성 고위험 초기 유방암의 수술 후 보조요법
14.5 생식세포 BRCA 돌연변이 HER2 음성 전이성 유방암의 치료
14.6 생식세포 BRCA 돌연변이 전이성 췌장 선암종의 일차 유지요법
14.7 HRR 유전자 돌연변이 전이성 거세저항성 전립선암
14.8 Abiraterone 및 Prednisone 또는 Prednisolone과 병용한 BRCA 돌연변이 전이성 거세저항성 전립선암 치료
16 공급/보관 및 취급 방법
17 환자 상담 정보
- *
- 전체 처방 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1 적응증 및 용법
1.1 BRCA 변이 진행성 난소암의 일차 유지요법
Lynparza는 백금 기반 일차 화학요법에 완전 또는 부분 반응을 보이는 유해하거나 유해할 것으로 의심되는 생식세포 또는 체세포 BRCA 변이가 있는 진행성 상피성 난소암, 난관암 또는 원발성 복막암 성인 환자의 유지요법에 사용된다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.2 Bevacizumab과 병용한 HRD 양성 진행성 난소암의 일차 유지요법
Lynparza는 백금 기반 일차 화학요법에 완전 또는 부분 반응을 보이고 다음 중 하나로 정의되는 상동 재조합 결핍(HRD) 양성 상태와 연관된 진행성 상피성 난소암, 난관암 또는 원발성 복막암 성인 환자의 유지요법에 bevacizumab과 병용투여 된다:
-
- •
- 유해하거나 유해할 것으로 의심되는 BRCA 변이, 그리고/또는
- •
- 유전체 불안정성.
Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.3 BRCA 변이 재발성 난소암의 유지요법
Lynparza는 백금 기반 화학요법에 완전 또는 부분 반응을 보이는 유해하거나 유해할 것으로 의심되는 생식세포 또는 체세포 BRCA 변이가 있는 재발성 상피성 난소암, 난관암 또는 원발성 복막암 성인 환자의 유지요법에 사용된다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.4 생식세포 BRCA 변이 HER2 음성 고위험 초기 유방암의 수술 후 보조요법
Lynparza는 수술 전 또는 수술 후 화학요법을 받은 유해하거나 유해할 것으로 의심되는 gBRCAm human epidermal growth factor receptor 2 (HER2) 음성 고위험 초기 유방암 성인 환자의 수술 후 보조요법에 사용된다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.5 생식세포 BRCA 변이 HER2 음성 전이성 유방암
Lynparza는 수술 전, 수술 후 또는 전이 단계에서 화학요법을 받은 유해하거나 유해할 것으로 의심되는 gBRCAm, HER2 음성 전이성 유방암 성인 환자의 치료에 사용된다. 호르몬 수용체(HR) 양성 유방암 환자는 이전에 내분비 요법을 받았거나 내분비 요법에 부적합한 것으로 간주되어야 한다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.6 생식세포 BRCA 변이 전이성 췌장 선암의 일차 유지요법
Lynparza는 백금 기반 일차 화학요법 요법을 최소 16주 이상 진행하는 동안 질병이 진행되지 않은 유해하거나 유해할 것으로 의심되는 gBRCAm 전이성 췌장 선암 성인 환자의 유지요법에 사용된다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.7 HRR 유전자 변이 전이성 거세저항성 전립선암
Lynparza는 enzalutamide 또는 abiraterone 치료 후 진행된 유해하거나 유해할 것으로 의심되는 생식세포 또는 체세포 상동 재조합 복구(HRR) 유전자 변이 전이성 거세저항성 전립선암(mCRPC) 성인 환자의 치료에 사용된다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
1.8 아비라테론, 프레드니손 또는 프레드니솔론과 병용한 BRCA 변이 전이성 거세저항성 전립선암 치료
Lynparza는 유해하거나 유해할 것으로 의심되는 BRCA 변이(BRCAm) 전이성 거세저항성 전립선암(mCRPC) 성인 환자 치료에 아비라테론, 프레드니손 또는 프레드니솔론과 병용 투여된다. Lynparza에 대해 FDA 승인을 받은 동반진단법에 근거하여 치료 대상 환자를 선택한다[용법용량(2.1) 참조].
2 용량 및 투여
2.1 환자 선택
유전자 변이 검출을 위한 FDA 승인 테스트 정보는 http://www.fda.gov/companiondiagnostics에서 확인할 수 있습니다.
Lynparza 치료 대상 환자는 적응증, 바이오마커, 샘플 유형에 따라 유해 또는 유해 가능성이 있는 HRR 유전자 변이(BRCA 변이 포함) 또는 유전체 불안정성 여부를 기준으로 선택합니다(표 1).
|
||||
|
적응증 |
바이오마커 |
샘플 유형 |
||
|
종양 |
혈액 |
혈장 (ctDNA) |
||
|
Body Germline 또는 Somatic BRCAm 진행성 난소암의 1차 유지 치료 |
BRCA1m, BRCA2m |
X |
X |
|
|
Bevacizumab와 병용하여 HRD 양성 진행성 난소암의 1차 유지 치료 |
BRCA1m, BRCA2m 및/또는 유전체 불안정성 |
X |
||
|
Germline 또는 Somatic BRCAm 재발성 난소암의 유지 치료 |
BRCA1m, BRCA2m |
X |
X |
|
|
gBRCAm HER2 음성 고위험 조기 유방암의 보조 치료 |
gBRCA1m, gBRCA2m |
X |
||
|
gBRCAm HER2 음성 전이성 유방암 |
gBRCA1m, gBRCA2m |
X |
||
|
Germline BRCA 변이 전이성 췌장암의 1차 유지 치료 |
gBRCA1m, gBRCA2m |
X |
||
|
Germline 또는 Somatic HRR 유전자 변이 전이성 내분비 치료 저항성 전립선암 |
ATMm, BRCA1m, BRCA2m, BARD1m, BRIP1m, CDK12m, CHEK1m, CHEK2m, FANCLm, PALB2m, RAD51Bm, RAD51Cm, RAD51Dm, RAD54Lm |
X |
||
|
gBRCA1m, gBRCA2m |
X |
|||
|
ATMm, BRCA1m, BRCA2m |
X |
|||
|
BRCA-변이 전립선암 확산성 호르몬 감수성 암과 아비라테론 및 프레드니손 또는 프레드니솔론과 연합하여 사용 |
BRCA1m, BRCA2m |
X |
X |
X |
2.2 권장 용량
Lynparza의 권장 용량은 하루에 300mg를 경구로 매일 두 번 복용하며, 식사 여부에 관계없이 복용합니다.
환자가 Lynparza 복용을 빠뜨린 경우, 다음 복용 시간에 맞춰 복용하도록 환자에게 안내하십시오.
환자에게 정제를 통째로 삼키도록 안내하십시오. 정제를 씹거나 분쇄, 용해 또는 분할하지 마십시오.
BRCA 변이 고급 난소암의 1차 유지 치료
질병 진행, 용인할 수 없는 독성 또는 2년 치료 완료까지 치료를 계속합니다. 2년 후 완전한 반응(영상학적으로 질병 증거 없음)을 보이는 환자는 치료를 중단해야 합니다. 2년 후 질병 증거가 있는 환자 중 치료를 계속할 경우, 치료를 계속할 수 있습니다.
HRD 양성 고급 난소암의 1차 유지 치료, 베바시주맙과 연합하여 사용
질병 진행, 용인할 수 없는 독성 또는 2년 치료 완료까지 Lynparza 치료를 계속합니다. 2년 후 완전한 반응(영상학적으로 질병 증거 없음)을 보이는 환자는 치료를 중단해야 합니다. 2년 후 질병 증거가 있는 환자 중 치료를 계속할 경우, 치료를 계속할 수 있습니다.
Lynparza와 함께 사용할 경우, 베바시주맙의 권장 용량은 3주마다 15mg/kg입니다. 베바시주맙은 화학요법과 함께 주어지며 유지 치료로 총 15개월 동안 투여되어야 합니다. 자세한 정보는 베바시주맙의 처방 정보를 참조하십시오.
유전자 변이 BRCA 양성 고위험 조기 유방암의 보조 치료
총 1년 동안 치료를 계속하거나 질병 재발, 용인할 수 없는 독성 중 먼저 발생하는 경우 치료를 계속합니다. 호르몬 수용체 양성 HER2 음성 유방암에 대해 Lynparza를 투여하는 환자는 현재의 임상 실천 지침에 따라 호르몬 요법을 동시에 계속해서 투여해야 합니다.
유전자 변이 BRCA 양성 난소암, 유전자 변이 BRCA 양성 HER2 음성 유방암, 유전자 변이 BRCA 양성 췌장선 암 및 HRR 유전자 변이 전립선암의 재발성
질병 진행 또는 용인할 수 없는 독성이 발생할 때까지 치료를 계속합니다:
-
- •
- 유전자 변이 BRCA 양성 난소암의 유지 치료.
- •
- 유전자 변이 BRCA 양성 HER2 음성 유방암.
- •
- 유전자 변이 BRCA 양성 췌장선 암의 1차 유지 치료.
- •
- HRR 유전자 변이 전립선암.
유전자 변이 BRCA 양성 전립선암, 아비라테론 및 프레드니손 또는 프레드니솔론과 연합하여 사용
질병 진행 또는 용인할 수 없는 독성이 발생할 때까지 치료를 계속합니다.
Lynparza와 함께 사용할 경우, 아비라테론의 권장 용량은 하루에 1000mg 경구로 한 번 복용합니다. 아비라테론은 프레드니손 또는 프레드니솔론 5mg를 하루에 두 번 경구로 복용해야 합니다. 용량 정보는 아비라테론의 처방 정보를 참조하십시오.
mCRPC 환자는 동시에 골반골수호르몬 방출 호르몬(GnRH) 아날로그를 병용하거나 양측 고환 절제술을 시행해야 합니다.
2.3 부작용에 대한 용량 조정
부작용을 관리하기 위해 치료 중단 또는 용량 감소를 고려하십시오. 권장 용량 감소는 하루에 250mg를 두 번 복용하는 것입니다.
더욱 용량 감소가 필요한 경우, 하루에 200mg를 두 번 복용하도록 감량하십시오.
2.4 강한 또는 중간 CYP3A 억제제와 동시 사용 시 용량 조정
Lynparza와 강한 또는 중간 CYP3A 억제제를 동시에 사용하지 마십시오.
동시 사용을 피할 수 없는 경우, Lynparza 용량을 다음과 같이 감량하십시오:
- •
- 강한 CYP3A 억제제와 동시 사용 시 하루에 100mg를 두 번 복용하십시오.
- •
- 중간 CYP3A 억제제와 동시 사용 시 하루에 150mg를 두 번 복용하십시오.
억제제가 중단된 후 3~5배의 소실기 반감기 동안 Lynparza 용량을 이전에 시작한 CYP3A 억제제 투여량으로 재개하십시오 [자세한 내용은 약물 상호작용 (7.2) 및 임상 약리학 (12.3)을 참조하십시오].
2.5 신장 기능 장애에 대한 용량 조정
중간 신장 기능 장애
중간 신장 기능 장애 환자(CLcr 31-50 mL/min)의 경우, Lynparza 용량을 하루에 200mg를 두 번 경구로 감량하십시오 [자세한 내용은 특정 인구에서의 사용 (8.6) 및 임상 약리학 (12.3)을 참조하십시오].
3 제형 및 함량
정제:
- •
- 150 mg: 녹색에서 녹색/회색을 띄는 타원형의 양면이 볼록한 필름코팅정제로서, 한 면에는 ‘OP150’이 음각되어 있고 반대면은 평평합니다.
- •
- 100 mg: 노란색에서 짙은 노란색을 띄는 타원형의 양면이 볼록한 필름코팅정제로서, 한 면에는 ‘OP100’이 음각되어 있고 반대면은 평평합니다.
4 금기 사항
없음.
5 경고 및 주의사항
5.1 골수형성이상증후군/급성골수성백혈병
Lynparza로 치료받은 환자에서 골수형성이상증후군(MDS)/급성골수성백혈병(AML)이 발생했으며 일부 사례는 치명적이었습니다.
임상 연구에서 승인된 적응증에 따라 단독요법 또는 병용요법으로 Lynparza를 투여받은 다양한 BRCAm, gBRCAm, HRR 유전자 변이 또는 HRD 양성 암 환자 2219명 중 MDS/AML의 누적 발생률은 약 1.2%(26/2219)였습니다. [이상반응 (6.1) 참조]. 이 중 54%(14/26)는 치명적인 결과를 초래했습니다. MDS/AML이 발생한 환자에서 Lynparza의 중간 치료 기간은 약 2년(범위: < 6개월 ~ > 4년)이었습니다. 이 환자들은 모두 이전에 백금계 약물 및/또는 방사선 요법을 포함한 기타 DNA 손상 약물로 화학요법을 받은 적이 있습니다.
새로 진단된 진행성 BRCAm 난소암 환자를 대상으로 한 SOLO1 연구에서 업데이트된 분석 결과, MDS/AML 발생률은 Lynparza를 투여받은 환자에서 1.9%(5/260), 위약을 투여받은 환자에서 0.8%(1/130)였습니다. 새로 진단된 HRD 양성 진행성 난소암 환자를 대상으로 한 PAOLA-1 연구에서 MDS/AML 발생률은 Lynparza를 투여받은 환자에서 1.6%(4/255), 대조군에서 2.3%(3/131)였습니다.
BRCAm 백금 민감성 재발성 난소암 환자를 대상으로 한 SOLO2 연구에서 MDS/AML 발생률은 Lynparza를 투여받은 환자에서 8%(15/195), 위약을 투여받은 환자에서 4%(4/99)였습니다. MDS/AML 진단 전 Lynparza 치료 기간은 0.6년에서 4.5년이었습니다.
이전 화학요법으로 인한 혈액학적 독성에서 회복될 때까지 Lynparza를 시작하지 마십시오(≤1등급). 기저치와 이후 매월 혈구감소증에 대한 전체 혈구 수를 모니터링하여 치료 중 임상적으로 유의한 변화가 있는지 확인하십시오. 장기간 혈액학적 독성이 있는 경우 Lynparza를 중단하고 회복될 때까지 매주 혈구수를 모니터링하십시오. 4주 후에도 수치가 1등급 이하로 회복되지 않으면 골수 분석 및 세포유전학 검사를 위한 혈액 샘플을 포함하여 추가 조사를 위해 환자를 혈액학자에게 의뢰하십시오. MDS/AML이 확인되면 Lynparza를 중단하십시오.
5.2 폐렴
Lynparza 단독요법으로 다양한 암 환자 2901명을 등록한 임상 연구에서 [이상반응(6.1) 참조] 치명적인 사례를 포함한 폐렴 발생률은 0.8%(24/2901)였습니다. 환자에게 호흡곤란, 기침, 발열과 같은 새로운 호흡기 증상이나 악화된 증상 또는 방사선학적 이상이 발생하는 경우 Lynparza 치료를 중단하고 즉시 증상의 원인을 평가하십시오. 폐렴이 확인되면 Lynparza 치료를 중단하고 환자를 적절히 치료하십시오.
5.3 정맥 혈전색전증
중증 또는 치명적인 폐색전증(PE)을 포함한 정맥 혈전색전증(VTE)이 Lynparza로 치료받은 환자에서 발생했습니다. [이상반응(6.1) 참조]
전이성 거세 저항성 전립선암 환자(N=1180)를 대상으로 한 무작위 위약 대조 임상 연구(PROfound 및 PROpel) 2건의 통합 데이터에서 Lynparza를 투여받은 환자의 8%에서 VTE가 발생했으며, 이 중 6%는 폐색전증이었습니다. 대조군에서는 VTE가 2.5% 발생했으며 이 중 1.5%는 폐색전증이었습니다.
정맥 혈전증 및 폐색전증의 임상 징후와 증상에 대해 환자를 모니터링하고 임상적으로 장기 항응고 요법을 포함하여 의학적으로 적절하게 치료하십시오.
5.4 배아-태아 독성
Lynparza는 작용기전과 동물 연구 결과를 바탕으로 임신부에게 투여 시 태아에게 해를 줄 수 있습니다. 동물 생식 연구에서 기관 형성 기간 동안 랫드에 olaparib을 투여하면 사람에게 권장되는 1일 2회 300mg 용량을 투여받은 환자에서보다 낮은 노출에서 기형발생 및 배아-태아 독성을 유발했습니다. 임신부에게 태아에 대한 잠재적 위험과 임신 손실의 잠재적 위험을 알리십시오. 가임기 여성에게 Lynparza 투여 중 및 최종 투여 후 6개월 동안 효과적인 피임법을 사용하도록 조언하십시오. 유전독성 및 동물 생식 연구 결과를 바탕으로 가임기 여성 파트너가 있거나 임신한 남성 환자에게 Lynparza 투여 중 및 최종 투여 후 3개월 동안 효과적인 피임법을 사용하도록 조언하십시오. [특정 집단에서의 사용 (8.1, 8.3) 참조]
6 ADVERSE REACTIONS
다음의 이상반응은 라벨의 다른 부분에서 논의되고 있습니다:
- •
- 골수형성이상증후군/급성골수성백혈병 [경고 및 주의사항 (5.1)을 참조]
- •
- 폐렴 [경고 및 주의사항 (5.2)를 참조]
- •
- 정맥 혈전색전증 [경고 및 주의사항 (5.3)을 참조]
6.1 임상시험 경험
임상시험은 매우 다양한 조건에서 실시되므로 약물의 임상시험에서 관찰된 이상반응 발생률을 다른 약물의 임상시험에서 발생률과 직접 비교할 수 없으며, 실제 실무에서의 발생률을 반영하지 않을 수 있습니다.
달리 명시되지 않는 한, 경고 및 주의사항에 설명된 데이터는 Lynparza 단일제로 투여 받은 2,901명의 환자에 노출된 것을 반영합니다. 이 중 2,135명 환자는 5개의 대조군, 무작위배정, 시험(SOLO-1, SOLO-2, OlympiAD, POLO, 및 PROfound)에서 1일 2회 300mg 정제 용량에 노출되었으며, 766명 환자는 다른 시험에서 1일 2회 400mg 캡슐 용량에 노출되어 안전성 분석을 실시했습니다. 이 2,901명의 환자 외에도, 경고 및 주의사항의 특정 절에는 Lynparza와 아비라테론(n=398)을 병용한 PROpel 시험에서 관찰된 이상반응이 포함되어 있습니다. 전이성 거세 저항성 전립선암 환자는 모두 동시에 안드로겐 박탈 요법(ADT) 또는 이전의 양측 고환절제술을 받았습니다.
이 통합된 안전성 집단에서 56%의 환자는 6개월 이상 노출되었고 28%의 환자는 1년 이상 Lynparza에 노출되었습니다.
이 통합된 안전성 집단에서 10% 이상의 환자에서 가장 흔하게 관찰된 이상반응은 구역(60%), 피로(55%), 빈혈(36%), 구토(32%), 설사(24%), 식욕감퇴(22%), 두통(16%), 미각장애(15%), 기침(15%), 호중구감소증(14%), 호흡곤란(14%), 어지럼증(12%), 소화불량(12%), 백혈구감소증(11%), 혈소판감소증(10%)였습니다.
BRCA 변이 진행성 난소암에 대한 1차 유지요법
SOLO-1
BRCA 변이 진행성 난소암 환자에서 1차 백금 기반 화학요법 이후 Lynparza의 유지요법의 안전성은 SOLO-1 [임상시험 정보 (14.1)를 참조]에서 조사되었습니다. 환자는 Lynparza 정제 300mg을 1일 2회 경구 투여(n=260) 또는 위약(n=130)을 질병 진행 또는 허용 불가능한 독성이 나타날 때까지 받았습니다. Lynparza를 투여받은 환자의 중앙 연구 치료 기간은 25개월, 위약 투여 환자는 14개월이었습니다.
Lynparza를 투여받은 환자 중 52%는 모든 등급의 이상반응으로 인해 투여 중단이, 28%는 모든 등급의 이상반응으로 인해 용량 감소가 있었습니다. Lynparza 투여 중단 또는 용량 감소의 가장 흔한 원인은 빈혈(23%), 구역(14%), 구토(10%)였습니다. 이상반응으로 인한 중단은 Lynparza 투여 환자의 12%에서 발생했으며, 가장 흔한 중단 원인은 피로(3.1%), 빈혈(2.3%), 구역(2.3%)이었습니다.
표 2 및 3은 SOLO-1에서 이상반응 및 실험실적 이상을 요약합니다.
|
||||
|
이상반응 |
Lynparza 정제 n=260 |
위약 n=130 |
||
|
전체 등급 (%) |
3-4등급 (%) |
전체 등급 (%) |
3-4등급 (%) |
|
|
위장관계 이상반응 |
||||
|
구역 |
77 |
1 |
38 |
0 |
|
복통† |
45 6 부작용 |
2 |
35 |
1 |
|
구토 |
40 |
0 |
15 |
1 |
|
설사‡ |
37 |
3 |
26 |
0 |
|
변비 |
28 |
0 |
19 |
0 |
|
소화불량 |
17 |
0 |
12 |
0 |
|
구내염§ |
11 |
0 |
2 |
0 |
|
전신 장애 및 투여부위 상태 |
||||
|
피로¶ |
67 |
4 |
42 |
2 |
|
혈액 및 림프계 장애 |
||||
|
빈혈 |
38 |
21 |
9 |
2 |
|
중성구감소증# |
17 |
6 |
7 |
3 |
|
백혈구감소증Þ |
13 |
3 |
8 |
0 |
|
혈소판감소증ß |
11 |
1 |
4 |
2 |
|
감염 및 기생충 침습 |
||||
|
상기도 감염/인플루엔자/비인두염/기관지염 |
28 |
0 |
23 |
0 |
|
요로감염à |
13 |
1 |
7 |
0 |
|
신경계 장애 |
||||
|
미각 장애 |
26 |
0 |
4 |
0 |
|
현기증 |
20 |
0 |
15 |
1 |
|
대사 및 영양 장애 |
||||
|
식욕 감퇴 |
20 |
0 |
10 |
0 |
|
호흡기, 흉부 및 종격 장애 |
||||
|
호흡곤란è |
15 |
0 |
6 |
0 |
린파자 투여 환자의 10% 미만에서 발생한 임상적으로 유의한 이상반응은 혈중 크레아티닌 증가(8%), 림프구감소증(6%), 정맥혈전색전증(3%), 과민반응(2%), 골수형성이상증후군/급성골수성백혈병(1.9%), 피부염(1%) 및 평균적혈구용적 증가(0.4%)였습니다.
|
실험실 지표* |
린파자 정제 n†=260 |
위약 n†=130 |
||
|
등급 1-4 (%) |
등급 3-4 (%) |
등급 1-4 (%) |
등급 3-4 (%) |
|
|
혈색소 감소 |
87 |
19 |
63 |
2 |
|
평균적혈구용적 증가 |
87 |
– |
43 |
– |
|
백혈구 감소 |
70 |
7 |
52 |
1 |
|
림프구 감소 |
67 |
14 |
29 |
5 |
|
절대 호중구 감소 |
51 |
9 |
38 |
6 |
|
혈소판 감소 |
35 |
1 |
20 |
2 |
|
혈청 크레아티닌 증가 |
34 |
0 |
18 |
0 |
베바시주맙과 병용 시 HRD-양성 진행성 난소암에 대한 일차 유지 치료
PAOLA-1
린파자와 베바시주맙의 병용에 의한 이상반응은 백금 기반 화학요법 및 베바시주맙를 포함하는 일차 치료 후 진행성 난소암에 대한 유지 치료의 PAOLA-1 연구에서 조사되었다 [임상시험 자료(14.2)]. 이는 이중맹검 위약 대조 연구로 802명의 환자가 무작위로 린파자 300 mg 1일 2회와 베바시주맙 복합군(n=535) 또는 위약과 베바시주맙 복합군(n=267)에 배정되었고, 질병 진행 또는 치료 불내성 시까지 치료받았다. 린파자/베바시주맙 병용군의 린파자 치료 기간 중위수는 17.3개월, 베바시주맙 치료 기간 중위수는 11개월이었다.
1명의 환자에서 동시다발 폐렴과 재생불량빈혈로 인한 치명적 이상반응이 발생하였다. 31%의 환자에서 중대한 이상반응이 발생했으며, 5% 이상에서 발생한 중대한 이상반응으로는 고혈압(19%), 빈혈(17%) 등이 있었다.
54%의 환자에서 어떤 등급의 이상반응으로 인한 투약 중단이 발생했고, 41%의 환자에서 용량 감량이 있었다.
린파자/베바시주맙 병용군에서 가장 흔한 투약 중단 이유는 빈혈(21%), 구역(7%), 구토(3%), 피로(3%) 등이었고, 가장 흔한 용량 감량 이유는 빈혈(19%), 구역(7%), 피로(4%) 등이었다.
20%의 환자에서 이상반응으로 인한 투약 중단이 있었으며, 가장 흔한 중단 이유는 빈혈(4%)과 구역(3%)이었다.
10% 이상에서 나타난 린파자/베바시주맙 병용군의 이상반응(위약/베바시주맙군 대비 5% 이상 발생 기준)은 구역(53%), 피로(53%), 빈혈(41%), 림프구감소증(24%), 구토(22%), 설사(18%), 호중구감소증(18%), 백혈구감소증(18%), 요로감염(15%), 두통(14%) 등이었다.
표 4와 5에 PAOLA-1 연구에서의 이상반응 및 실험실적 이상값을 각각 정리하였다.
|
이상반응 |
린파자/베바시주맙 n=535 |
위약/베바시주맙 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
1-4등급 (%) |
3-4등급 (%) |
1-4등급 (%) |
3-4등급 (%) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
전신 및 투여부위 이상 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
피로(권태감 포함)† |
53 |
5 |
32 |
1.5 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
소화기계 이상 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
구역 |
53 |
2.4 |
22 |
0.7 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
구토 |
22 |
1.7 |
11 |
1.9 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
혈액 및 림프계 이상 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
빈혈‡ |
41 |
17 |
10 |
0.4 린파구감소증§ 24% 7% 9% 1.1% 백혈구감소증¶ 18% 1.9% 10% 1.5% 10% 미만의 환자에서 발생한 임상적으로 관련 있는 이상반응은 미각이상(8%), 호흡곤란(8%), 구내염(5%), 소화불량(4.3%), 홍조(3%), 현기증(2.6%), 과민반응(1.7%) 및 MDS/AML(0.7%)이었습니다. 정맥혈전색전증은 린파자/베바시주맙 투여 환자군(5%)에서 위약/베바시주맙 투여 환자군(1.9%)보다 더 흔하게 발생했습니다.
BRCA 돌연변이 재발성 난소암의 유지 요법 SOLO-2 BRCA 돌연변이 재발성 난소암 환자의 유지 요법으로서 린파자의 안전성은 SOLO-2 연구에서 조사되었습니다 [임상시험 결과 14.3 참조]. 환자는 린파자 정제 300mg을 1일 2회 경구 투여(n=195) 또는 위약(n=99)을 질병 진행 또는 허용 불가능한 독성이 나타날 때까지 투여 받았습니다. 린파자 투여군의 중앙 치료 기간은 19.4개월, 위약군은 5.6개월이었습니다. 린파자 복용 환자 중 45%에서 모든 등급의 이상반응으로 인한 투여 중단이 발생했고, 27%에서 이상반응으로 인한 용량 감량이 있었습니다. 린파자 투여 중단 또는 감량의 가장 흔한 원인은 빈혈(22%), 호중구감소증(9%), 그리고 피로/무력증(8%)이었습니다. 린파자 투여군 환자의 11%가 이상반응으로 투여를 중단했습니다. 표 6과 7은 SOLO-2 연구에서 발생한 이상반응과 검사치 이상을 요약한 것입니다.
Lynparza 복용 환자의 20% 미만에서 발생한 임상적으로 중요한 이상반응으로는 호중구감소증(19%), 기침(18%), 백혈구감소증(16%), 저마그네슘혈증(14%), 혈소판감소증(14%), 현기증(13%), 소화불량(11%), 크레아티닌 증가(11%), MDS/AML(8%), 부종(8%), 발진(6%), 정맥 혈전색전증(5%), 및 림프구감소증(1%)이 있었습니다.
린파자 투여 환자의 10% 미만에서 발생한 임상적으로 유의한 이상반응은 기침(9.2%), 림프구감소증(7%), 소화불량(6%), 상복부 통증(4.9%), 발진(4.9%), 호흡곤란(4.2%), 혈소판감소증(4.2%), 크레아티닌 증가(2%), 과민반응(0.9%), 정맥혈전색전증(0.5%), 피부염(0.5%), 평균 적혈구 용적 증가(0.2%), 골수 형성이상 증후군/급성 골수성 백혈병(0.2%) 등이었습니다.
|
평균 적혈구 용적 증가‡
|
67
|
0
|
4.8
|
0
|
헤모글로빈 감소
|
65
|
8
|
31
|
0.9
|
백혈구 감소
|
64
|
5
|
42
|
0.7
|
절대 호중구 수 감소
|
39
|
7
|
27
|
1.1 Germline BRCA 돌연변이 HER2-음성 전이성 유방암 OlympiAD Lynparza의 안전성은 전이성 질환에 대해 이전에 최대 2회의 화학요법을 받은 HER2-음성 전이성 유방암 환자를 대상으로 OlympiAD 시험에서 평가되었습니다 [임상시험 정보 (14.5)]. 환자들은 Lynparza 정제 300 mg을 1일 2회 경구 투여(n=205) 또는 의료진의 선택에 따른 화학요법제(카페시타빈, 에리뷰린 또는 비노렐빈, n=91)를 질병이 진행되거나 허용할 수 없는 독성이 나타날 때까지 투여받았습니다. 연구 치료 기간의 중앙값은 Lynparza를 투여받은 환자에서 8.2개월, 화학요법을 투여받은 환자에서 3.4개월이었습니다. Lynparza를 투여받은 환자 중 35%에서 모든 등급의 이상반응으로 인한 투약 중단이 있었고, 25%에서 이상반응으로 인한 투여량 감소가 있었습니다. 이상반응으로 인한 투약 중단은 Lynparza 투여 환자의 5%에서 발생했습니다. 표 10과 11은 OlympiAD에서 이상반응과 실험실 검사치 이상을 요약한 것입니다.
Lynparza 복용 환자의 20% 미만에서 발생한 임상적으로 유의한 이상반응으로는 기침(18%), 식욕감소(16%), 혈소판감소증(11%), 미각장애(9%), 림프구감소증(8%), 소화불량(8%), 현기증(7%), 구내염(7%), 상복부 통증(7%), 발진(5%), 혈청 크레아티닌 증가(3%), 피부염(1%) 및 정맥혈전색전증(1%)이 있습니다.
Lynparza 복용 환자의 10% 미만에서 발생한 임상적으로 유의한 부작용으로는 기침(9%), 상복부 통증(7%), 혈중 크레아티닌 증가(7%), 현기증(7%), 두통(7%), 소화불량(5%), 백혈구감소증(5%), 정맥 혈전색전증(3%), 과민반응(2%), 림프구감소증(2%) 등이 있었습니다.
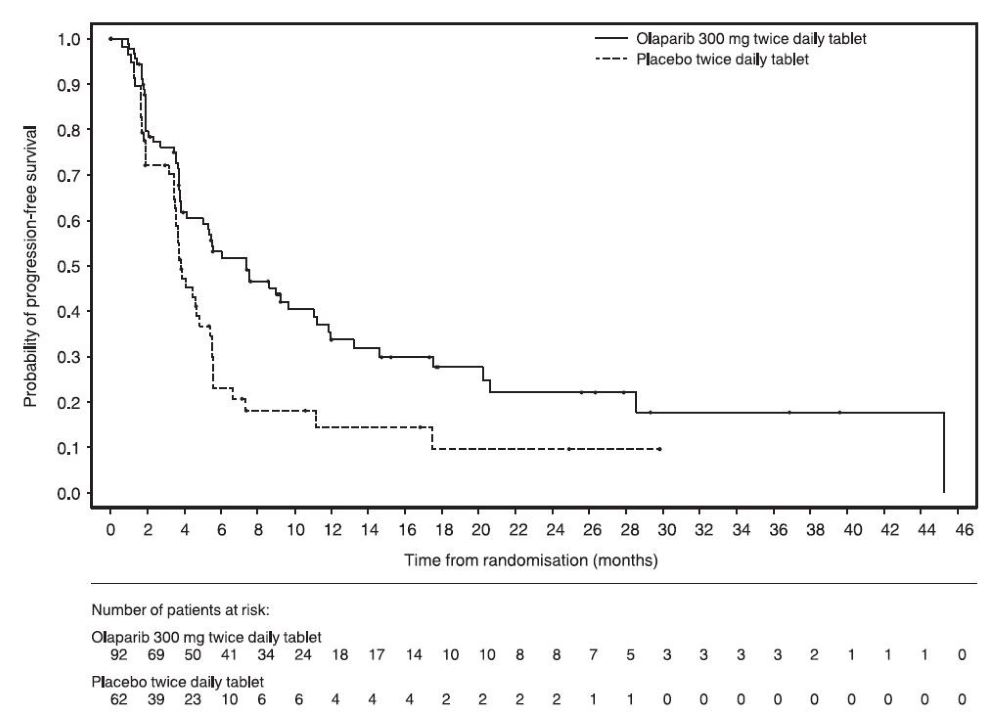
HRR 유전자 돌연변이 전이성 불응성 전립선암 PROfound Lynparza 단일 요법의 안전성은 이전에 enzalutamide 또는 abiraterone을 투여받은 후 진행된 mCRPC 및 HRR 유전자 돌연변이 환자를 대상으로 한 PROfound 연구에서 평가되었습니다 [임상 연구 (14.7)]. 이 연구는 무작위 배정, 공개 라벨, 다기관 연구로, 386명의 환자가 Lynparza 정제 300mg을 1일 2회 경구 투여(n=256)하거나 또는 investigator’s 선택에 따라 enzalutamide 또는 abiraterone acetate(n=130)를 질병 진행 또는 허용 불가능한 독성이 발생할 때까지 투여받았습니다. Lynparza를 투여받은 환자 중 62%는 6개월 이상, 20%는 1년 이상 노출되었습니다. 치명적 이상반응이 Lynparza 치료 환자의 4%에서 발생했습니다. 이에는 폐렴(1.2%), 심폐 부전(0.4%), 흡인 폐렴(0.4%), 장 게실증(0.4%), 패혈증 쇼크(0.4%), Budd-Chiari 증후군(0.4%), 급사(0.4%), 급성 심부전(0.4%) 등이 포함되었습니다. 중대한 이상반응이 Lynparza 투여 환자의 36%에서 발생했습니다. 가장 흔한 중대한 이상반응(≥2%)은 빈혈(9%), 폐렴(4%), 폐색전증(2%), 피로/무력증(2%), 요로 감염(2%) 등이었습니다. Lynparza 투여 환자의 45%에서 이상반응으로 인한 투여 중단이 발생했으며, 22%에서 이상반응으로 인한 용량 감소가 있었습니다. Lynparza의 투여 중단을 가장 많이 일으킨 이상반응은 빈혈(25%) 및 혈소판 감소증(6%)이었고, 용량 감소를 가장 많이 일으킨 이상반응은 빈혈(16%)이었습니다. 이상반응으로 인한 투여 중단이 Lynparza 환자의 18%에서 발생했으며, Lynparza 투여 중단을 가장 많이 일으킨 이상반응은 빈혈(7%)이었습니다. 표 14와 15는 각각 PROfound 연구에서의 이상반응과 실험실 검사치 이상을 요약합니다.
Lynparza를 투여받은 환자의 10% 미만에서 발생한 임상적으로 유의한 이상반응에는 호중구감소증(9%), 정맥혈전증(7%), 어지러움(7%), 미각 장애(7%), 소화불량(7%), 두통(6%), 폐렴(5%), 구내염(5%), 발진(4%), 혈중 크레아티닌 증가(4%), 간질성 폐렴(2%), 상복부 통증(2%), 과민반응(1%) 등이 있었다.
BRCA 돌연변이 전이성 거세저항성 전립선암에 대한 아비라테론 및 프렛니손 또는 프레드니솔론 병용 치료 PROpel Lynparza와 아비라테론 및 프레드니손 또는 프레드니솔론 병용요법의 안전성은 최초 치료군 mCRPC 환자를 대상으로 한 PROpel 연구에서 평가되었다 [임상시험 정보 (14.8) 참조]. 환자들은 Lynparza 정제 300 mg 1일 2회 경구 투여 및 아비라테론 정제 1000 mg 1일 1회 투여군(Lynparza/아비라테론)(n=398) 또는 위약 및 아비라테론 1000 mg 1일 1회 투여군(위약/아비라테론)(n=396)으로 무작위 배정되었다. 이 두 군 모두 프레드니손 또는 프레드니솔론 5 mg 1일 2회를 병용 투여하였다. 치명적인 이상반응은 환자의 6%에서 발생하였는데, COVID-19(3%) 및 폐렴(0.5%)이 포함되었다. 중대한 이상반응은 환자의 39%에서 발생하였다. 2% 이상의 환자에서 보고된 중대한 이상반응으로는 빈혈(6%), COVID-19(6%), 폐렴(4.5%), 폐색전증(3.5%), 요로감염(3%) 등이 있었다. 이상반응으로 인한 Lynparza 영구 투여 중단은 Lynparza/아비라테론 병용군의 16%에서 발생하였다. Lynparza 영구 투여 중단을 초래한 가장 흔한 이상반응은 빈혈(4.3%) 및 폐렴(1.5%)이었다. 린파자(Lynparza) 투여 중단은 린파자와 아비라테론 병용군 환자의 48%에서 발생했습니다. 린파자 투여 중단을 요하는 가장 흔한 (>2%) 이상반응은 빈혈(16%), 코로나19 감염(6%), 피로(3.5%), 구역(2.8%), 폐색전증(2.3%), 설사(2.3%) 등이었습니다. 린파자의 용량 감량은 린파자와 아비라테론 병용군 환자의 21%에서 발생했습니다. 린파자의 용량 감량을 요하는 가장 흔한 (>2%) 이상반응은 빈혈(11%) 및 피로(2.5%)였습니다. 린파자/아비라테론을 투여한 환자에서 가장 흔한 이상반응(≥10%)은 빈혈(48%), 피로(38%), 구역(30%), 설사(19%), 식욕감소(16%), 림프구감소증(14%), 복통(13%), 현기증(14%) 등이었습니다. 표 16과 17은 각각 PROpel 연구에서 이상반응과 실험실적 이상 결과를 요약합니다.
Lynparza 및 아비라테론 투여 환자에서 10% 미만으로 발생한 임상적으로 관련 있는 부작용은 두통(9%), 정맥혈전색전증(8%), 발진(7%), 미각장애(6%), 급성 신부전(3%) 및 구내염(2.5%)이었습니다.
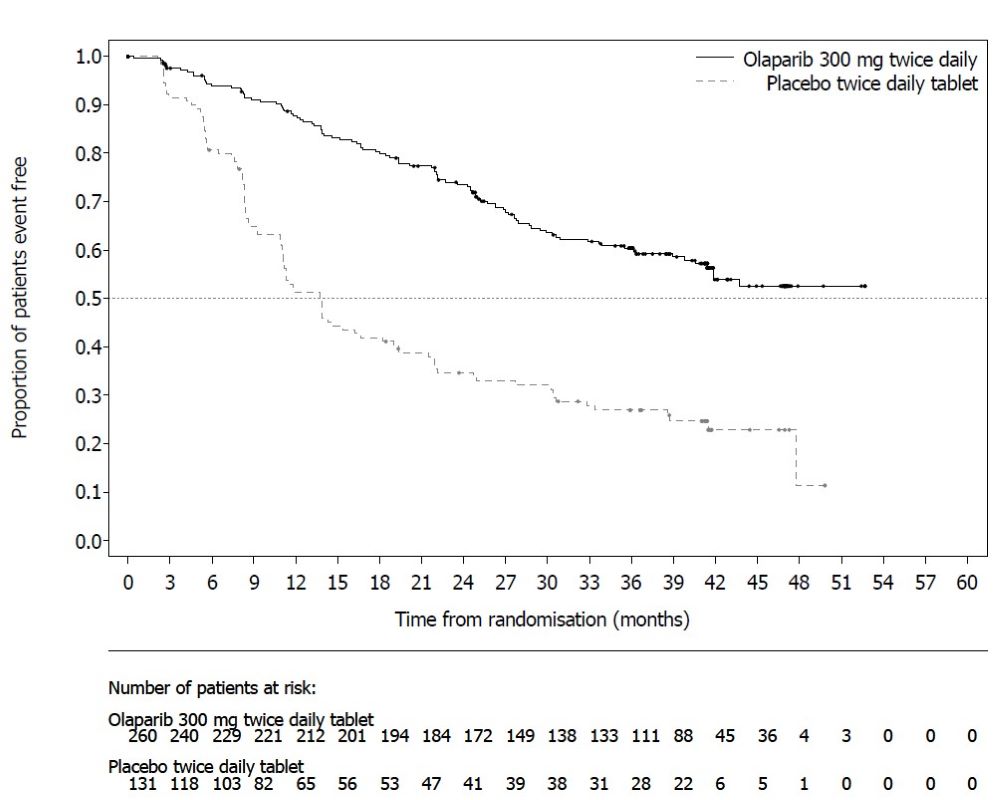
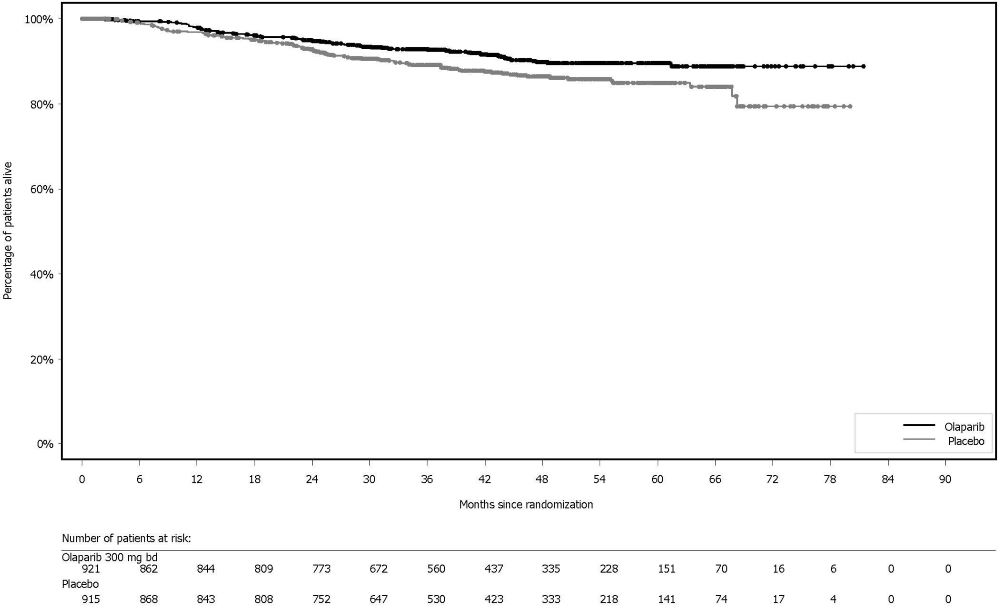
6.2 시판 후 경험다음 부작용은 Lynparza의 시판 후 사용 중 확인되었습니다. 이러한 반응은 불확실한 규모의 인구집단에서 자발적으로 보고되었기 때문에 항상 빈도를 신뢰성 있게 추정하거나 약물 노출과의 인과관계를 확립할 수는 없습니다. 면역계 장애: 혈관부종을 포함한 과민반응. 피부 및 피하조직 장애: 홍반성 구진, 발진, 피부염. 7 약물 상호작용7.2 Lynparza에 대한 다른 약물의 영향강력한 및 중등도 CYP3A 억제제 CYP3A 억제제와 병용 투여 시 olaparib 농도가 증가하여 부작용 위험이 높아질 수 있습니다 [임상 약리학 (12.3) 참조]. 강력하거나 중등도 CYP3A 억제제와 병용 투여를 피하십시오. 강력하거나 중등도 억제제를 반드시 병용 투여해야 하는 경우 Lynparza 용량을 감량하십시오 [용법 및 용량 (2.4) 참조]. 강력한 및 중등도 CYP3A 유도제 강력하거나 중등도 CYP3A 유도제와 병용 시 olaparib 노출이 감소하여 Lynparza 효능이 감소할 수 있습니다 [임상 약리학 (12.3) 참조]. 강력하거나 중등도 CYP3A 유도제와 병용 투여를 피하십시오. 8 특정 집단에서의 사용8.1 임신위험 요약 동물 연구 결과와 작용기전에 근거할 때 [임상 약리학 (12.1) 참조], Lynparza는 임신한 여성에게 투여 시 태아에게 해를 끼칠 수 있습니다. 임신 중 Lynparza 사용에 대한 약물 관련 위험을 알려줄 수 있는 가용한 데이터는 없습니다. 동물 생식 연구에서, 기관 형성기 동안 임신한 랫트에게 olaparib 투여 시, 권장 인간 용량인 1일 2회 300 mg보다 낮은 노출 수준에서 기형 유발 및 배아-태아 독성을 유발했습니다 (데이터 참조). 임신한 여성에게 태아에 대한 잠재적 위험과 임신 손실의 잠재적 위험을 알려주십시오. 해당 집단에 대한 주요 선천성 결함 및 유산의 추정 배경 위험은 알려져 있지 않습니다. 미국 일반 인구에서 주요 선천성 결함의 추정 배경 위험은 2-4%이며, 임상적으로 인지된 임신에서 자연 유산의 위험은 약 15-20%입니다. 데이터동물 데이터 암컷 랫트에서 수행한 생식능 및 초기 배아 발달 연구에서, olaparib를 교미 전 14일부터 임신 6일까지 경구 투여한 결과, 15 mg/kg/day 용량(권장 용량에서 인체 노출(AUC0-24h)의 약 7%에 해당하는 모체 전신 노출)에서 착상 후 손실이 증가했습니다. 배아-태아 발달 연구에서, 임신한 랫트는 기관 형성기 동안 olaparib 0.05 및 0.5 mg/kg/day의 경구 용량을 투여 받았습니다. 0.5 mg/kg/day 용량(권장 용량에서 인체 노출(AUC0-24h)의 약 0.18%에 해당하는 모체 전신 노출)은 착상 후 손실 증가와 눈(anophthalmia, microphthalmia), 척추/늑골(여분의 늑골 또는 골화 중심, 융합되거나 없는 척추궁, 늑골 및 흉골), 두개골(융합된 exoccipital) 및 횡격막(탈장)의 주요 기형을 포함한 배아-태아 독성을 유발했습니다. 추가 이상 또는 변이로는 불완전하거나 없는 골화(척추/흉골, 늑골, 사지)와 척추/흉골, 골반대, 폐, 흉선, 간, 요관 및 제대 동맥의 기타 소견이 포함되었습니다. 위에서 언급한 눈, 늑골 및 요관의 일부 소견은 더 낮은 발생률로 olaparib 0.05 mg/kg/day 용량에서 관찰되었습니다. 8.2 수유위험 요약 olaparib가 모유에 존재하는지, 또는 수유 중인 영아나 모유 생성에 미치는 영향에 대한 데이터는 없습니다. Lynparza로 인한 수유 중인 영아에서의 중대한 이상반응 가능성 때문에, 수유부에게 Lynparza 치료 중 및 마지막 용량 투여 후 1개월 동안 수유하지 않도록 권고하십시오. 8.3 가임기 여성 및 남성Lynparza는 임신한 여성에게 투여 시 태아에게 해를 끼칠 수 있습니다 [특정 집단에서의 사용 (8.1) 참조]. 임신 검사 Lynparza 치료 시작 전에 가임기 여성의 임신 상태를 확인하십시오. 피임 여성 가임기 여성에게 Lynparza 치료 중 및 마지막 용량 투여 후 6개월 동안 효과적인 피임법을 사용하도록 권고하십시오. 남성 유전독성 및 동물 생식 연구 결과에 근거하여, 가임기 여성 파트너가 있거나 임신한 남성 환자에게 치료 중 및 Lynparza의 마지막 용량 투여 후 3개월 동안 효과적인 피임법을 사용하도록 권고하십시오. 남성 환자에게 Lynparza 치료 중 및 마지막 용량 투여 후 3개월 동안 정자를 기증하지 않도록 권고하십시오 [특정 집단에서의 사용 (8.1) 및 비임상 독성학 (13.1) 참조]. 8.5 고령자 사용진행성 고형암으로 Lynparza 단독요법을 받은 2901명의 환자 중, 680명(23%)은 65세 이상이었고, 이 중 206명(7%)은 75세 이상이었습니다. 13명(0.4%)의 환자는 85세 이상이었습니다. 진행성 고형암으로 Lynparza 정제 300mg을 베바시주맙과 병용하여 1일 2회 경구 투여받은 535명의 환자(PAOLA-1) 중, 204명(38%)은 65세 이상이었고, 이 중 31명(6%)은 75세 이상이었습니다. 진행성 고형암으로 Lynparza 정제 300mg을 아비라테론 및 프레드니손 또는 프레드니솔론과 병용하여 1일 2회 경구 투여받은 398명의 환자(PROpel) 중, 268명(67%)은 65세 이상이었고, 이 중 95명(24%)은 75세 이상이었습니다. 이러한 환자와 더 젊은 환자 사이에 Lynparza의 안전성 또는 유효성에 있어 전반적인 차이는 관찰되지 않았습니다. 8.6 신장애경증 신장애 환자(Cockcroft-Gault로 추정한 CLcr 51~80 mL/min)에서는 용량 조절이 권장되지 않습니다. 중등도 신장애 환자(CLcr 31~50 mL/min)에서는 Lynparza 용량을 1일 2회 200mg으로 감량하십시오 [용법용량 (2.5) 참조]. 중증 신장애 또는 말기 신장 질환(CLcr ≤30 mL/min) 환자에 대한 데이터는 없습니다 [임상 약리학 (12.3) 참조]. 8.7 간장애경증 또는 중등도 간장애(Child-Pugh 분류 A 및 B) 환자에서 시작 용량 조절은 필요하지 않습니다. 중증 간장애(Child-Pugh 분류 C) 환자에 대한 데이터는 없습니다 [임상 약리학 (12.3) 참조]. 11 제품 설명Olaparib은 poly (ADP-ribose) polymerase (PARP) 억제제입니다. 화학명은 4-[(3-{[4-(cyclopropylcarbonyl)piperazin-1-yl]carbonyl}-4-fluorophenyl)methyl]phthalazin-1(2H)-one입니다. Lynparza의 실험식은 C24H23FN4O3이고 상대 분자량은 434.46입니다. 다음과 같은 화학 구조를 가지고 있습니다: Olaparib은 결정성 고체이며, 비대칭이고 생리학적 pH 범위에서 pH에 무관하게 낮은 용해도를 보입니다. 경구용 Lynparza (olaparib) 정제는 100 mg 또는 150 mg의 olaparib을 함유하고 있습니다. 정제 코어의 비활성 성분으로는 copovidone, mannitol, colloidal silicon dioxide, sodium stearyl fumarate가 있습니다. 정제 코팅은 hypromellose, polyethylene glycol 400, titanium dioxide, ferric oxide yellow, ferrosoferric oxide(150 mg 정제에만 해당)로 구성되어 있습니다. 12 임상약리12.1 작용기전Olaparib은 PARP1, PARP2, PARP3을 포함한 poly (ADP-ribose) polymerase (PARP) 효소의 억제제입니다. PARP 효소는 DNA 전사 및 DNA 복구와 같은 정상적인 세포 기능에 관여합니다. Olaparib은 in vitro에서 선택된 종양 세포주의 성장을 억제하고 단독요법 또는 platinum 기반 화학요법 후 인간 암의 mouse xenograft 모델에서 종양 성장을 감소시키는 것으로 나타났습니다. BRCA1/2, ATM 또는 DNA 손상의 상동 재조합 복구(HRR)에 관여하는 다른 유전자에 결함이 있는 세포주 및 mouse 종양 모델에서 olaparib 치료 후 세포독성 및 항종양 활성이 증가되었으며 platinum 반응과 상관관계가 있었습니다. In vitro 연구에 따르면 olaparib 유도 세포독성은 PARP 효소 활성 억제 및 PARP-DNA 복합체 형성 증가와 관련될 수 있으며, 이로 인해 DNA 손상 및 암세포 사멸이 발생합니다. 전립선암 모델에서 PARP1은 androgen receptor (AR) 활성 조절에 기여하는 것으로 나타났습니다. Olaparib과 AR 억제제의 병용은 in vitro에서 세포독성을 나타냈으며 mouse xenograft 모델에서 항종양 활성을 보였습니다. 12.2 약력학심장 전기생리학 Olaparib이 심장 재분극에 미치는 영향은 300 mg 단회 투여 후 119명의 환자와 300 mg 1일 2회 반복 투여 후 109명의 환자에서 평가되었습니다. Olaparib이 QT 간격에 임상적으로 관련된 영향은 관찰되지 않았습니다. 12.3 약동학Olaparib의 AUC (area under the curve)는 25 mg에서 450 mg (권장 용량의 0.08~1.5배)의 단회 투여 후 용량에 비례하여 증가하였고, 최고 농도(Cmax)는 동일한 용량 범위에서 용량에 비해 약간 덜 증가하였습니다. Olaparib은 시간 의존적 약동학을 보였으며, 300 mg을 1일 2회 투여할 때 정상 상태에서 1.8의 AUC 평균 축적 비율이 관찰되었습니다. 300 mg 단회 투여 후 olaparib의 평균(CV%) Cmax는 5.4 μg/mL (32%), AUC는 39.2 μg*h/mL (44%)입니다. 300 mg을 1일 2회 투여할 때 olaparib의 평균 정상 상태 Cmax는 7.6 μg/mL (35%), AUC는 49.2 μg*h/mL (44%)입니다. 흡수 Olaparib 경구 투여 후 혈장 최고 농도에 도달하는 시간 중앙값은 1.5 시간입니다. 음식의 영향 고지방 및 고칼로리 식사(800-1000 kcal, 칼로리의 50%가 지방으로 구성)와 olaparib을 병용 투여하면 olaparib 흡수 속도(tmax가 2.5시간 지연)는 느려졌지만, olaparib 흡수 정도는 유의하게 변화하지 않았습니다(평균 AUC가 약 8% 증가). 분포 Lynparza 300 mg 단회 투여 후 olaparib의 평균(± 표준편차) 겉보기 분포 용적은 158 ± 136 L입니다. In vitro에서 olaparib의 단백 결합은 약 82%입니다. 소실 Lynparza 300 mg 단회 투여 후 olaparib의 평균(± 표준편차) 말단 혈장 반감기는 14.9 ± 8.2 시간이고 겉보기 혈장 청소율은 7.4 ± 3.9 L/h입니다. 대사 Olaparib은 in vitro에서 cytochrome P450 (CYP) 3A에 의해 대사됩니다. 방사성 표지된 olaparib을 여성 환자에게 경구 투여한 후, 혈장에서 순환하는 방사능의 70%가 변하지 않은 olaparib이었습니다. 소변과 대변에서 방사능의 15%와 6%가 각각 변하지 않은 약물로 설명되는 광범위한 대사를 겪었습니다. 대사의 대부분은 산화 반응에 기인하며, 생성된 성분 중 다수는 이후 글루쿠론산 또는 황산 포합을 겪습니다. 배설 방사성 표지된 olaparib의 단회 투여 후 7일 수집 기간 동안 투여된 방사능의 86%가 회수되었으며, 44%는 소변으로, 42%는 대변으로 회수되었습니다. 대부분의 물질은 대사체로 배설되었습니다. 특정 집단 신장애 환자 신장애 시험에서 olaparib을 경증 신장애 환자(Cockcroft-Gault 공식으로 정의된 CLcr=51-80 mL/min; n=13)에게 투여했을 때 정상 신기능 환자(CLcr ≥81 mL/min; n=12)와 비교하여 평균 AUC가 24%, Cmax가 15% 증가했고, 중등도 신장애 환자(CLcr=31-50 mL/min; n=13)에게 투여했을 때는 각각 44%와 26% 증가했습니다. Olaparib의 혈장 단백 결합 정도와 크레아티닌 청소율 사이의 관계에 대한 증거는 없었습니다. 중증 신장애 또는 말기 신장 질환(CLcr ≤30 mL/min) 환자에 대한 데이터는 없습니다. 간장애 환자 간장애 시험에서 olaparib을 경증 간장애 환자(Child-Pugh 분류 A; n=10)에게 투여했을 때 정상 간기능 환자(n=13)와 비교하여 평균 AUC가 15% 증가하고 평균 Cmax가 13% 증가했으며, 중등도 간장애 환자(Child-Pugh 분류 B; n=8)에게 투여했을 때는 평균 AUC가 8% 증가하고 평균 Cmax는 13% 감소했습니다. 간장애는 olaparib의 단백 결합에 영향을 미치지 않으므로 총 혈장 노출은 유리 약물의 대표였습니다. 중증 간장애 환자(Child-Pugh 분류 C)에 대한 데이터는 없습니다. 약물 상호작용 연구 임상 연구 CYP3A 억제제: Itraconazole (강력한 CYP3A 억제제)과 병용 투여 시 olaparib의 Cmax가 42%, AUC가 170% 증가했습니다. Fluconazole (중등도 CYP3A 억제제)과 병용 투여 시 olaparib의 Cmax가 14%, AUC가 121% 증가할 것으로 예측됩니다. CYP3A 유도제: Rifampicin (강력한 CYP3A 유도제)과 병용 투여 시 olaparib의 Cmax가 71%, AUC가 87% 감소했습니다. Efavirenz (중등도 CYP3A 유도제)와 병용 투여 시 olaparib의 Cmax가 31%, AUC가 60% 감소할 것으로 예측됩니다. In vitro 연구 CYP 효소: Olaparib은 CYP3A의 억제제이자 유도제이며 CYP2B6의 유도제입니다. Olaparib은 사람에서 약한 CYP3A 억제제로 예측됩니다. UGT 효소: Olaparib은 UGT1A1의 억제제입니다. 수송체: Olaparib은 BCRP, OATP1B1, OCT1, OCT2, OAT3, MATE1 및 MATE2K의 억제제입니다. Olaparib은 배출 수송체 P-gp의 기질이자 억제제입니다. Olaparib의 P-gp 유도 가능성은 평가되지 않았습니다. 13 비임상 독성학13.1 발암성, 변이원성, 생식능력 장애Olaparib에 대한 발암성 연구는 수행되지 않았습니다. Olaparib은 포유류 Chinese hamster ovary (CHO) 세포의 in vitro 염색체 이상 분석과 in vivo 랫드 골수 소핵 분석에서 clastogenic했습니다. 이러한 clastogenicity는 olaparib의 일차 약리학에서 비롯된 게놈 불안정성과 일치하며 인간에 대한 잠재적 genotoxicity를 나타냅니다. Olaparib은 박테리아 역돌연변이(Ames) 시험에서 돌연변이를 유발하지 않았습니다. 생식능력 연구에서 암컷 랫드는 교배 전 최소 14일부터 임신 첫 주까지 0.05, 0.5, 15 mg/kg/day의 용량으로 경구 olaparib을 투여받았습니다. 15 mg/kg/day 용량까지 교배 및 생식능력에 대한 부작용은 없었습니다(권장 용량에서 인체 노출(AUC0-24h)의 약 7%에 해당하는 모체 전신 노출). 수컷 생식능력 연구에서 olaparib은 최소 70일간의 olaparib 투여 후 최대 40 mg/kg/day의 경구 용량에서 랫드의 교배 및 생식능력에 영향을 미치지 않았습니다(권장 용량에서 인체 노출(AUC0-24h)의 약 5%에 해당하는 전신 노출). 14 CLINICAL STUDIES14.1 첫 번째 라인 유지 치료 BRCA-돌연변이 진행성 난소암린파자의 효능은 SOLO-1(NCT01844986)에서 평가되었다. 이는 무작위 배정(2:1), 이중 맹검, 위약 대조, 다기관 시험으로, 첫 번째 라인 백금 기반 화학요법 이후 BRCA-돌연변이 진행성 난소암, 나팔관암, 또는 원발성 복막암 환자를 대상으로 했다. 환자들은 린파자 정제 300mg을 경구 투여하거나 위약을 무작위로 배정받았다. 치료는 최대 2년 또는 질병 진행 또는 허용할 수 없는 독성이 있을 때까지 계속되었다. 단, 2년 후에도 질병의 징후가 있으며 치료 의료진의 의견으로 지속 치료로부터 더 이익을 얻을 수 있다고 판단되는 환자는 2년 이상 치료받을 수 있었다. 무작위 배정은 첫 번째 라인 백금 기반 화학요법에 대한 반응(완전 반응 또는 부분 반응)을 기준으로 계층화되었다. 주요 유효성 평가 변수는 고형암 반응 평가 기준(RECIST) 1.1판에 따라 조사자가 평가한 무진행 생존기간(PFS)이었다. 총 391명의 환자가 무작위 배정되었고, 260명은 린파자, 131명은 위약을 투여받았다. 린파자 투여 환자의 중앙 연령은 53세(범위: 29~82세), 위약군은 53세(범위: 31~84세)였다. 환자 활동 정도(ECOG PS)는 린파자군에서 77%, 위약군에서 80%가 0점이었다. 전체 환자의 82%가 백인이었고, 36%가 미국 또는 캐나다에서 등록되었으며, 82%가 가장 최근 백금 기반 요법에 완전 반응을 보였다. 대다수 환자(n=389)는 생식계 BRCA 돌연변이(gBRCAm)를 가지고 있었고, 2명의 환자는 체세포 BRCA 돌연변이(sBRCAm)를 가지고 있었다. SOLO-1에서 무작위 배정된 391명의 환자 중 386명은 사후 또는 전향적으로 Myriad BRACAnalysis 검사를 받았고, 383명에서 유해 또는 의심 유해 gBRCAm 상태가 확인되었다. 253명은 린파자군, 130명은 위약군으로 무작위 배정되었다. SOLO-1에서 무작위 배정된 391명 중 2명은 조직 검사 Foundation Medicine에 의해 sBRCAm로 확인되었다. SOLO-1에서 린파자는 위약에 비해 조사자 평가 PFS에서 통계적으로 유의한 개선을 보였다. 맹검 독립 검토 결과도 일치했다. PFS 분석 시점에서 전체 생존(OS) 데이터는 아직 성숙하지 않았다(21%의 환자가 사망). 유효성 결과는 표 18 및 그림 1과 같다.
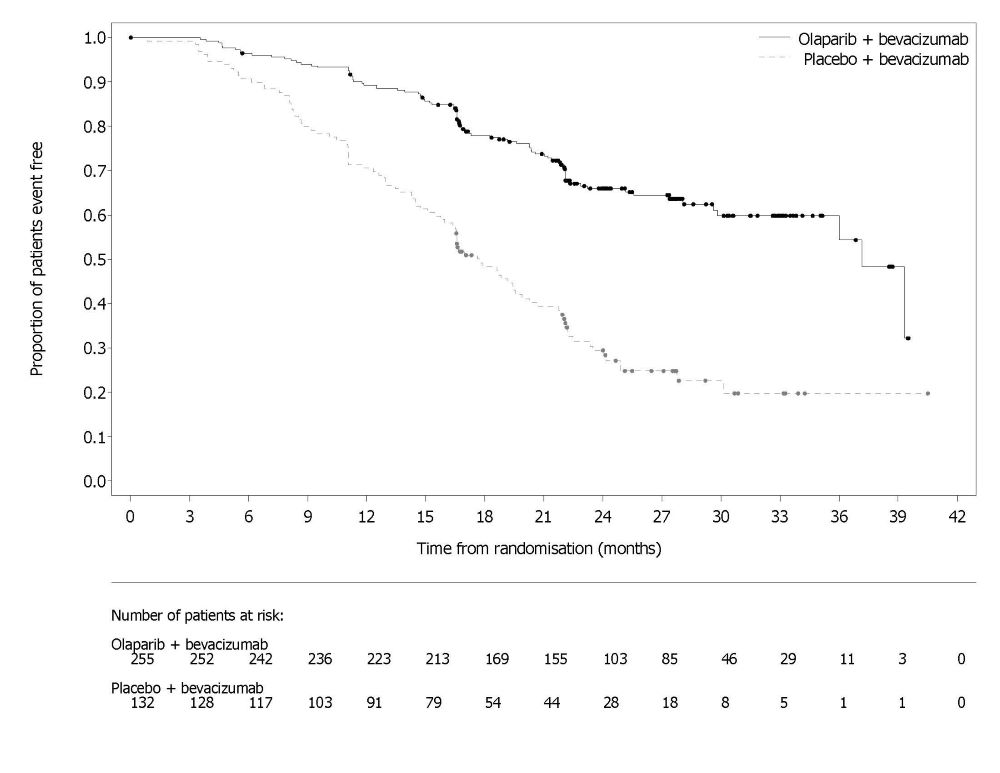
그림 1 조사자 평가 무진행 생존기간 Kaplan-Meier 곡선 – SOLO-1 14.2 음성 진행성 난소암 1차 유지요법으로서 Bevacizumab과 병용투여PAOLA-1 (NCT02477644)은 1차 백금 기반 화학요법과 bevacizumab 후 진행성 상피성 난소암, 난관암 또는 원발성 복막암 유지요법으로 bevacizumab과 병용한 Lynparza의 효능을 위약/bevacizumab과 비교한 무작위 이중 맹검 위약 대조 다기관 시험이었습니다. 무작위 배정은 1차 치료 결과(세포감소술의 시기 및 결과와 백금 기반 화학요법에 대한 반응)와 전향적 현지 검사로 결정된 tBRCAm 상태에 따라 층화되었습니다. 이용 가능한 모든 임상 검체는 Myriad myChoice® CDx로 후향적으로 검사되었습니다. 환자는 완전한 수술적 절제로 인한 질병의 증거가 없거나, 1차 백금 함유 화학요법과 bevacizumab 완료 후 완전 반응(CR) 또는 부분 반응(PR)을 보인 환자여야 했습니다. 환자는 bevacizumab 15mg/kg을 3주마다 투여하면서 Lynparza 정제 300mg을 1일 2회 경구 투여하는 군(n=537)과 위약/bevacizumab 투여군(n=269)에 2:1로 무작위 배정되었습니다. 환자는 유지요법 환경에서 bevacizumab을 계속 투여했고, 마지막 화학요법 투여 후 최소 3주에서 최대 9주 이내에 Lynparza 투여를 시작했습니다. Lynparza 치료는 기저 질환의 진행 또는 허용할 수 없는 독성이 나타날 때까지 최대 2년간 지속되었습니다. 지속적인 치료로 추가적인 이점을 얻을 수 있다고 담당 의사가 판단한 환자는 2년 이상 치료를 받을 수 있었습니다. Bevacizumab으로 치료하는 기간은 화학요법과 함께 투여한 기간과 유지요법으로 투여한 기간을 포함하여 총 15개월까지였습니다. 주요 효능 평가 지표는 RECIST 버전 1.1에 따라 평가된 연구자 평가 PFS였습니다. 추가 효능 평가 지표는 전체 생존기간(OS)이었습니다. 전체 분석 대상(ITT) 집단에서 PFS에 통계적으로 유의한 차이가 관찰되었습니다. 알려진 HRD 상태의 환자에서 PFS 및 OS에 대한 계획된 탐색적 분석이 수행되었습니다. HRD 음성 종양 환자(277/806; 34%)의 PFS 위험비(HR)는 1.00 (95% CI: 0.75, 1.34)이었고 OS HR은 1.18 (95% CI: 0.87, 1.60)로, 임상적 이점이 주로 HRD 양성 집단에서 관찰된 결과에 기인한 것으로 나타났습니다. HRD 양성 종양 환자에서의 효능 결과는 표 19, 그림 2 및 그림 3에 요약되어 있습니다. 무작위 배정 후 Myriad myChoice® HRD Plus 종양 검사를 사용하여 확인된 HRD 양성 종양 환자 387명(48%) 중 양쪽 군 모두에서 연령 중앙값은 58세(범위 32-82세)였습니다. 양쪽 군 모두에서 87%의 환자의 원발 종양 유형은 난소암이었습니다. 95%는 장액성 조직학적 유형이었습니다. ECOG 수행 능력 점수는 75%의 환자에서 0, 24%의 환자에서 1이었습니다. 모든 환자는 1차 백금 기반 요법과 bevacizumab을 받았습니다. 선별 시 1차 치료 결과는 초기 종양감소술에서 완전한 대식세포 절제술로 질병의 증거가 없는 환자(36%), 간격 종양감소술에서 완전한 대식세포 절제술로 질병/CR의 증거가 없는 환자(양쪽 군 29%), 절제가 불완전한 환자(초기 또는 간격 종양감소술에서) 또는 종양감소술을 받지 않은 환자(양쪽 군 16%)에서 질병/CR의 증거가 없는 환자, 부분 반응 환자(양쪽 군 19%)였습니다. Lynparza/bevacizumab군 환자의 62%, 위약/bevacizumab군 환자의 58%는 유해한 BRCA 변이가 있는 종양이 있었습니다. 환자는 수술 결과에 제한을 받지 않았으며, 67%는 초기 또는 간격 종양감소술에서 완전한 세포감소술을 받았고 33%는 잔류 대식종양을 가지고 있었습니다.
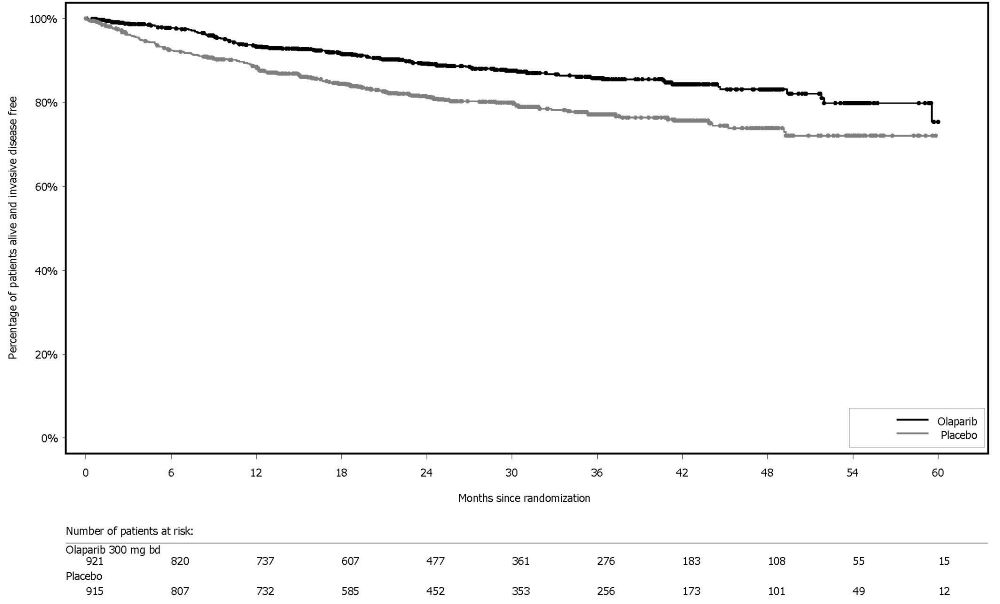
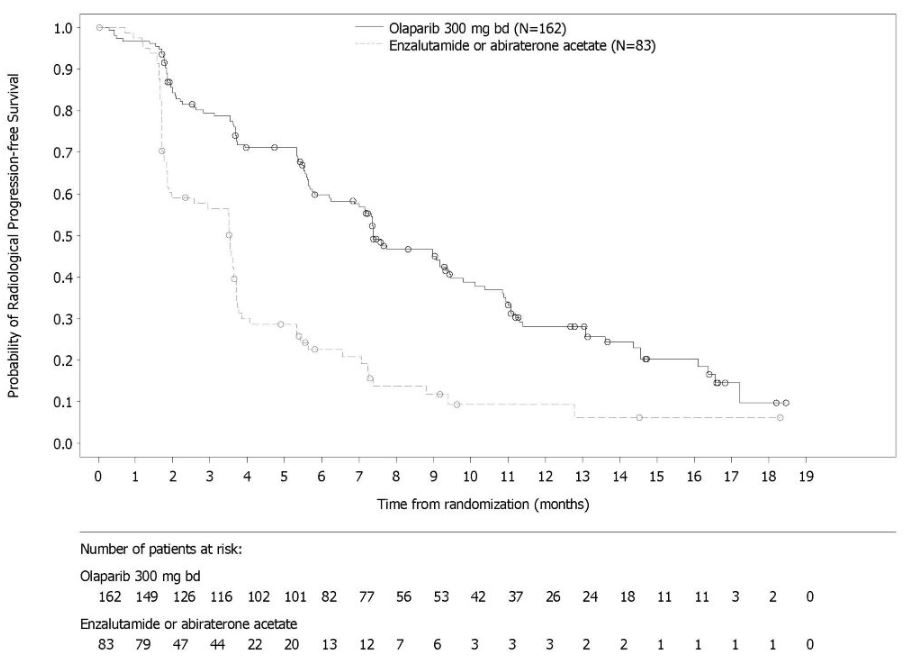
그림 10 PROfound – 코호트 A의 BICR 평가 rPFS에 대한 Kaplan-Meier 곡선 이전에 taxane 요법을 받은 환자와 받지 않은 환자에 대한 rPFS의 탐색적 분석에서 일관된 결과가 관찰되었으며, Myriad BRACAnalysis CDx 분석으로 확인된 germline-BRCA 돌연변이 환자와 Foundation Medicine F1CDx 분석으로 확인된 BRCA 돌연변이 환자에서도 일관된 결과가 관찰되었다. 그림 11 PROfound – 코호트 A의 전체 생존율에 대한 Kaplan-Meier 곡선 린파자 투여군의 HRR 돌연변이별 반응 데이터는 표 27에 제시되어 있다. 코호트 A와 B의 비교군에서 총 3명의 환자가 부분 반응을 나타냈는데, 1명은 ATM 단일 돌연변이 환자, 2명은 공동 발생 돌연변이 환자(PALB2+PPP2R2A 1명, CDK12+PALB2 1명)였다.
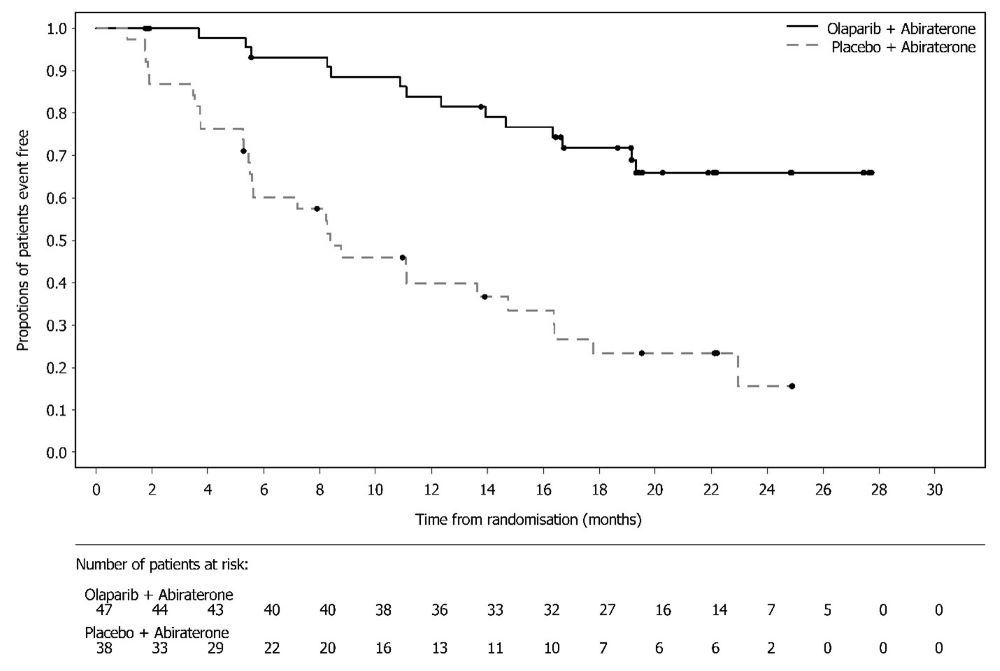
14.8 BRCA 돌연변이 전이성 거세 내성 전립선암에서 아비라테론과 프레드니손 또는 프레드니솔론과의 병용 치료린파자의 전이성 거세 내성 전립선암 치료 효과는 PROpel (NCT03732820), 무작위 배정, 이중 눈가림, 위약 대조, 다기관 연구에서 평가되었다. 이 연구에서는 전이성 거세 내성 전립선암 환자에서 린파자의 아비라테론 병용 요법과 아비라테론 단독요법을 비교 평가하였다. 환자(n=796명)는 린파자 정제 300mg을 1일 2회 경구 투여 및 아비라테론 1000mg 1일 1회와 병용(n=399명)하는 군 또는 위약과 아비라테론(n=397명)을 병용하는 군에 1:1 무작위 배정되었다. 모든 환자는 프레드니손 또는 프레드니솔론 5mg을 1일 2회 투여받았고, GnRH 유사체 또는 이전 양측 고환적출술을 받았다. 이전 아비라테론 치료 경험이 있는 환자는 제외되었다. 국소 또는 전이성 호르몬 민감성 전립선암(mHSPC)에서의 이전 도세탁셀 치료는 허용되었다. 무작위 배정 시 전이 부위 (골전이만, 내장 전이 또는 기타)와 mHSPC 단계에서의 도세탁셀 치료 여부(예 또는 아니오)로 층화되었다. 객관적인 방사선학적 질병 진행이 판정되기 전까지 또는 허용할 수 없는 독성이 발생하기 전까지 린파자 치료를 지속하였다. BRCA 유전자 돌연변이(BRCAm) 상태는 무작위 배정 후에 NGS 기반의 종양 조직 및 ctDNA 검사로 모두 평가되었다. FDA 승인 검사와 일치하는 BRCAm 분류 기준을 사용하여 환자의 체세포 또는 생식세포 돌연변이 상태에 대한 유해 및 의심 유해 돌연변이 여부를 판정하였다. 주요 유효성 평가변수는 RECIST 1.1판 및 전립선암 실무그룹 3 (PCWG3) (골전이) 기준에 따라 평가한 판정자 평가 방사선학적 무진행 생존기간(rPFS)이었다. 전체 생존기간(OS)은 추가 유효성 평가변수였다. 총 796명의 환자 중 85명(11%)이 ctDNA 검사(9%) 또는 종양 조직 검사(6%)에서 BRCAm 양성으로 판정되었다. 이 85명 중 중앙연령은 68세(범위 43-85세)였고, 67%가 65세 이상이었다. 72%가 백인, 22%가 아시아인, 2%가 흑인 또는 아프리카계 미국인이었다. ECOG 활동상태는 66%가 0등급, 34%가 1등급이었다. 25%가 이전에 mHSPC에서 도세탁셀 치료 받았다. 53%는 골전이만 있었고, 15%는 내장 전이, 32%는 기타 전이가 있었다. 린파자/아비라테론군이 위약/아비라테론군에 비해 전체 대상군(ITT)에서 통계적으로 유의한 rPFS 연장을 보였다. 총 711명의 BRCAm 음성 환자군에서의 탐색 분석에서는 rPFS 위험비 0.77 (95% CI: 0.63, 0.96), OS 위험비 0.92 (95% CI: 0.74, 1.14)로 ITT 군에서의 유의한 결과는 주로 BRCAm 양성 환자군 결과에 기인하는 것으로 나타났다. PROpel 연구의 85명 BRCAm 환자군에서의 탐색 분석 결과가 표 28, 그림 12 및 13에 요약되어 있다. BICR 평가 결과도 판정자 평가 rPFS 결과와 일치하였다.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||