의약품 제조업체: Pharmacyclics LLC (Updated: 2024-12-20)
처방 정보 하이라이트
IMBRUVICA® (ibrutinib) 캡슐, 경구용
IMBRUVICA® (ibrutinib) 정제, 필름 코팅, 경구용
IMBRUVICA® (ibrutinib) 경구용 현탁액
미국 최초 승인: 2013
최근 주요 변경 사항
| 경고 및 주의 사항, 간독성, 약물 유발 간 손상 포함 (5.7) | 2024년 5월 |
적응증 및 용법
용법 및 용량
금기 사항
없음 (4)
경고 및 주의 사항
- 출혈: 출혈을 모니터링하고 관리하십시오 (5.1).
- 감염: 환자의 발열 및 감염을 모니터링하고 즉시 평가하고 치료하십시오 (5.2).
- 심장 부정맥, 심부전 및 돌연사: 부정맥 및 심부전 증상을 모니터링하고 관리하십시오 (5.3).
- 고혈압: 혈압을 모니터링하고 치료하십시오 (5.4).
- 혈구감소증: 매달 완전 혈구 수를 확인하십시오 (5.5).
- 제2의 원발성 악성 종양: 피부암 및 기타 암종을 포함하여 환자에게 다른 악성 종양이 발생했습니다 (5.6).
- 간독성, 약물 유발 간 손상 포함: 치료 전반에 걸쳐 간 기능을 모니터링하십시오 (5.7).
- 종양 용해 증후군(TLS): 기준 위험을 평가하고 예방 조치를 취하십시오. TLS를 모니터링하고 치료하십시오 (5.8).
- 배아-태아 독성: 태아에게 해를 끼칠 수 있습니다. 임신 가능성이 있는 여성에게 태아에 대한 잠재적 위험을 알리고 효과적인 피임법을 사용하도록 조언하십시오 (5.9, 8.1, 8.3).
유해 반응
- B 세포 악성 종양 환자에서 가장 흔한 (≥30%) 유해 반응은 혈소판 감소증, 설사, 피로, 근골격계 통증, 호중구 감소증, 발진, 빈혈, 멍, 오심입니다 (6).
- cGVHD 성인 또는 소아 환자에서 가장 흔한 (≥20%) 유해 반응은 피로, 빈혈, 멍, 설사, 혈소판 감소증, 근골격계 통증, 발열, 근육 경련, 구내염, 출혈, 오심, 복통, 폐렴 및 두통입니다 (6).
의심되는 유해 반응을 보고하려면 Pharmacyclics(1-877-877-3536) 또는 FDA(1-800-FDA-1088 또는 www.fda.gov/medwatch)에 문의하십시오.
약물 상호작용
특정 환자군에서의 사용
환자 상담 정보 및 FDA 승인 환자 라벨링은 17번을 참조하십시오.
개정: 2024년 12월
목차
전문 정보: 목차*
1
적응증 및 용법
1.1
만성 림프구성 백혈병/소림프구성 림프종
1.2
17p 결손이 있는 만성 림프구성 백혈병/소림프구성 림프종
1.3
발덴스트롬 거대글로불린혈증
1.4
만성 이식편대숙주병
2
용법 및 용량
2.1
권장 용량
2.2
유해 반응에 대한 용량 조절
2.3
CYP3A 저해제와 병용 시 용량 조절
2.4
간 손상 환자에서의 용량 조절
3
제형 및 함량
4
금기
5
경고 및 주의사항
5.1
출혈
5.2
감염
5.3
심부정맥, 심부전 및 돌연사
5.4
고혈압
5.5
혈구감소증
5.6
제2의 원발성 악성종양
5.7
약물 유발 간 손상을 포함한 간독성
5.8
종양 용해 증후군
5.9
배아-태아 독성
6
유해 반응
6.1
임상 시험 경험
6.2
시판 후 경험
7
약물 상호작용
7.1
CYP3A 저해제가 이브루티닙에 미치는 영향
7.2
CYP3A 유도제가 이브루티닙에 미치는 영향
8
특정 환자군에서의 사용
8.1
임신
8.2
수유
8.3
생식 가능 연령 여성 및 남성
8.4
소아 사용
8.5
노인 사용
8.6
간 손상
8.7
혈장분리술
10
과량 투여
11
약물 설명
12
임상 약리학
12.1
작용 기전
12.2
약력학
12.3
약동학
13
비임상 독성학
13.1
발암성, 돌연변이 유발성, 생식능 저해
14
임상 연구
14.1
만성 림프구성 백혈병/소림프구성 림프종
14.2
발덴스트롬 거대글로불린혈증
14.3
만성 이식편대숙주병
16
포장 단위/보관 및 취급
17
환자 상담 정보
- *
- 전문 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1 적응증 및 사용법
1.2
17p 결손이 있는 만성 림프구성 백혈병/소림프구성 림프종
IMBRUVICA는 17p 결손이 있는 만성 림프구성 백혈병(CLL)/소림프구성 림프종(SLL) 성인 환자의 치료에 사용됩니다.
2 용법 및 투여
2.1
권장 용량
만성 림프구성 백혈병/소림프구성 림프종 및 발덴스트롬 마크로글로불린혈증
CLL/SLL 및 WM에 대한 IMBRUVICA의 권장 용량은 질병 진행 또는 용인할 수 없는 독성이 나타날 때까지 1일 1회 경구 420mg입니다.
CLL/SLL의 경우, IMBRUVICA는 단독 요법으로, 리툭시맙 또는 오비누투주맙 병용 요법으로, 또는 벤다무스틴 및 리툭시맙(BR) 병용 요법으로 투여할 수 있습니다.
WM의 경우, IMBRUVICA는 단독 요법으로 또는 리툭시맙 병용 요법으로 투여할 수 있습니다.
리툭시맙 또는 오비누투주맙과 IMBRUVICA를 병용 투여하는 경우, 같은 날 투여할 때는 IMBRUVICA를 리툭시맙 또는 오비누투주맙보다 먼저 투여하는 것을 고려하십시오.
만성 이식편대숙주병
cGVHD가 있는 12세 이상 환자의 경우 IMBRUVICA의 권장 용량은 1일 1회 경구 420mg이며, cGVHD가 있는 1세 이상 12세 미만 환자의 경우 1일 1회 경구 240mg/m2(최대 420mg까지)입니다. cGVHD 치료가 더 이상 필요하지 않은 경우, 개별 환자의 의학적 평가를 고려하여 IMBRUVICA 투여를 중단해야 합니다.
| 240 mg/m2를 달성하기 위한 권장 용량 | ||
| BSA*(m2) 범위 | 투여할 IMBRUVICA 캡슐/정제 용량(mg) | 투여할 IMBRUVICA 경구용 현탁액(70 mg/mL) 용량(mL) |
| > 0.3 ~ 0.4 | – | 1.2 mL |
| > 0.4 ~ 0.5 | – | 1.5 mL |
| > 0.5 ~ 0.6 | – | 1.9 mL |
| > 0.6 ~ 0.7 | – | 2.2 mL |
| > 0.7 ~ 0.8 | 210 mg | 2.6 mL |
| > 0.8 ~ 0.9 | 210 mg | 2.9 mL |
| > 0.9 ~ 1 | 210 mg | 3.3 mL |
| > 1 ~ 1.1 | 280 mg | 3.6 mL |
| > 1.1 ~ 1.2 | 280 mg | 4 mL |
| > 1.2 ~ 1.3 | 280 mg | 4.3 mL |
| > 1.3 ~ 1.4 | 350 mg | 4.6 mL |
| > 1.4 ~ 1.5 | 350 mg | 5 mL |
| > 1.5 ~ 1.6 | 350 mg | 5.3 mL |
| > 1.6 | 420 mg | 6 mL |
*BSA = 체표면적 (body surface area).
투여
IMBRUVICA는 매일 거의 같은 시간에 투여하십시오.
정제 또는 캡슐을 물 한 잔과 함께 삼키십시오. 캡슐을 열거나, 부수거나, 씹지 마십시오. 정제를 자르거나, 부수거나, 씹지 마십시오.
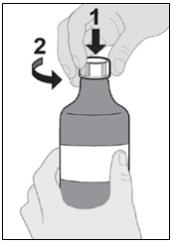
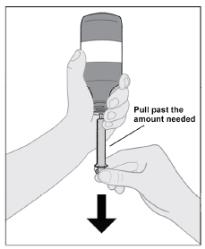
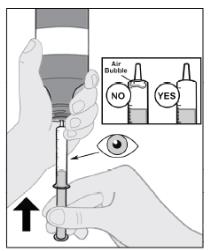
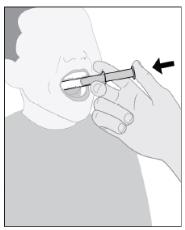
IMBRUVICA 경구 현탁액의 추가 투여 세부 정보는 사용 지침을 따르십시오.
예정된 시간에 IMBRUVICA 용량을 복용하지 않은 경우, 같은 날 가능한 한 빨리 복용하고 다음 날 정상 일정으로 돌아갈 수 있습니다. 놓친 용량을 보충하기 위해 IMBRUVICA를 추가로 복용하지 마십시오.
2.2
유해 반응에 대한 용량 조절
표 2에 나열된 유해 반응의 경우 IMBRUVICA 치료를 중단하십시오. 유해 반응이 1등급 또는 기준선(회복)으로 개선되면 권장 용량 조절을 따르십시오(표 2 참조).
| 유해 반응a,b | 발생 | CLL/SLL, WM 및 12세 이상 환자의 용량 조절 회복 후 cGVHD 시작 용량 = 420mg |
1세 이상 12세 미만 cGVHD 환자의 용량 조절 회복 후 시작 용량 = 240mg/m2 |
| 2등급 심부전 | 첫 번째 | 1일 280mg으로 재시작c | 1일 160mg/m2으로 재시작c |
| 두 번째 | 1일 140mg으로 재시작c | 1일 80mg/m2 으로 재시작c | |
| 세 번째 | IMBRUVICA 중단 | IMBRUVICA 중단 | |
| 3등급 심장 부정맥 | 첫 번째 | 1일 280mg으로 재시작c | 1일 160mg/m2으로 재시작c |
| 두 번째 | IMBRUVICA 중단 | IMBRUVICA 중단 | |
| 3등급 또는 4등급 심부전 4등급 심장 부정맥 |
첫 번째 | IMBRUVICA 중단 | IMBRUVICA 중단 |
| 기타 3등급 또는 4등급 비혈액학적 독성d 3등급 또는 4등급 호중구 감소증 감염 또는 발열 동반 4등급 혈액학적 독성 |
첫 번째 | 1일 280mg으로 재시작 | 1일 160mg/m2으로 재시작c |
| 두 번째 | 1일 140mg으로 재시작 | 1일 80mg/m2 으로 재시작c | |
| 세 번째 | IMBRUVICA 중단 | IMBRUVICA 중단 |
a [경고 및 주의사항 (5) 참조].
b 국립암연구소-유해사례 공통 용어 기준(NCI-CTCAE) 또는 만성 림프구성 백혈병 국제 워크숍(iwCLL) 기준에 따른 CLL/SLL의 혈액 독성에 대한 등급입니다.
c 치료 재개 전에 유익성과 위험성을 평가하십시오.
d 4등급 비혈액 독성의 경우, 치료 재개 전에 유익성과 위험성을 평가하십시오.
| 160 mg/m2 달성을 위한 권장 용량 | 80 mg/m2 달성을 위한 권장 용량 | |||
| BSA*(m2) 범위 | 투여할 IMBRUVICA 캡슐/정제 용량 (mg) | 투여할 IMBRUVICA 경구용 현탁액 (70 mg/mL) 용량 (mL) | 투여할 IMBRUVICA 캡슐/정제 용량 (mg) | 투여할 IMBRUVICA 경구용 현탁액 (70 mg/mL) 용량 (mL) |
| > 0.3 ~ 0.4 | – | 0.8 mL | – | 0.4 mL |
| > 0.4 ~ 0.5 | – | 1 mL | – | 0.5 mL |
| > 0.5 ~ 0.6 | – | 1.3 mL | – | 0.6 mL |
| > 0.6 ~ 0.7 | – | 1.5 mL | – | 0.7 mL |
| > 0.7 ~ 0.8 | 140 mg | 1.7 mL | 70 mg | 0.9 mL |
| > 0.8 ~ 0.9 | 140 mg | 1.9 mL | 70 mg | 1 mL |
| > 0.9 ~ 1 | 140 mg | 2.2 mL | 70 mg | 1.1 mL |
| > 1 ~ 1.1 | 140 mg | 2.4 mL | 70 mg | 1.2 mL |
| > 1.1 ~ 1.2 | 210 mg | 2.6 mL | – | 1.3 mL |
| > 1.2 ~ 1.3 | 210 mg | 2.9 mL | – | 1.4 mL |
| > 1.3 ~ 1.4 | 210 mg | 3.1 mL | – | 1.5 mL |
| > 1.4 ~ 1.5 | 210 mg | 3.3 mL | 140 mg | 1.7 mL |
| > 1.5 ~ 1.6 | 280 mg | 3.5 mL | 140 mg | 1.8 mL |
| > 1.6 | 280 mg | 4 mL | 140 mg | 2 mL |
*BSA = 체표면적 (body surface area).
2.3
CYP3A 저해제와 병용 시 용량 조절
권장 용량 조절은 아래에 설명되어 있습니다 [약물 상호작용 (7.1)] 참조:
| 환자군 | 병용 약물 | 권장 IMBRUVICA 용량 |
| B 세포 악성 종양 |
|
1일 1회 280mg
권장대로 용량 조절 [용법 및 용량 (2.2)] 참조. |
|
1일 1회 140mg
권장대로 용량 조절 [용법 및 용량 (2.2)] 참조. |
|
|
1일 1회 70mg
권장대로 용량 중단 [용법 및 용량 (2.2)] 참조. |
|
|
병용 투여하지 마십시오.
이러한 저해제를 단기간(예: 7일 이하의 항감염제) 사용하는 경우 IMBRUVICA를 중단하십시오. |
|
| cGVHD 환자 12세 이상 |
|
1일 1회 420mg
권장대로 용량 조절 [용법 및 용량 (2.2)] 참조. |
|
1일 1회 280mg
권장대로 용량 조절 [용법 및 용량 (2.2)] 참조. |
|
|
1일 1회 140mg
권장대로 용량 중단 [용법 및 용량 (2.2)] 참조. |
|
|
병용 투여하지 마십시오.
이러한 저해제를 단기간(예: 7일 이하의 항감염제) 사용하는 경우 IMBRUVICA를 중단하십시오. |
|
| cGVHD 환자 1세 이상 12세 미만 |
|
1일 1회 240mg/m2
권장대로 용량 조절 [용법 및 용량 (2.2)] 참조. |
|
1일 1회 160mg/m2 | |
|
1일 1회 80mg/m2 | |
|
병용 투여하지 마십시오.
이러한 저해제를 단기간(예: 7일 이하의 항감염제) 사용하는 경우 IMBRUVICA를 중단하십시오. |
CYP3A 저해제 중단 후, 이전 IMBRUVICA 용량으로 재개하십시오 [용법 및 용량 (2.1), 약물 상호작용 (7.1)].
2.4
간 손상 환자의 용량 조절
B 세포 악성 종양 성인 환자
경도 간 손상 환자(Child-Pugh 등급 A)의 권장 용량은 1일 140mg입니다.
중등도 간 손상 환자(Child-Pugh 등급 B)의 권장 용량은 1일 70mg입니다.
중증 간 손상 환자(Child-Pugh 등급 C)에게는 IMBRUVICA 사용을 피하십시오 [특정 환자군에서의 사용 (8.6), 임상 약리학 (12.3)].
cGVHD 환자
총 빌리루빈 수치가 정상 상한치의 1.5배 초과 3배 이하인 12세 이상 환자의 권장 용량은 1일 140mg입니다 (비간성 기원이 아니거나 길버트 증후군 때문이 아닌 경우).
총 빌리루빈 수치가 정상 상한치의 1.5배 초과 3배 이하인 1세 이상 12세 미만 환자의 권장 용량은 1일 80mg/m2입니다 (비간성 기원이 아니거나 길버트 증후군 때문이 아닌 경우).
총 빌리루빈 수치가 정상 상한치의 3배를 초과하는 환자에게는 IMBRUVICA 사용을 피하십시오 (비간성 기원이 아니거나 길버트 증후군 때문이 아닌 경우) [특정 환자군에서의 사용 (8.6), 임상 약리학 (12.3)].
3 제형 및 함량
캡슐:
70mg 캡슐은 검은색 잉크로 “ibr 70mg”라고 표시된 노란색 불투명 캡슐입니다.
140mg 캡슐은 검은색 잉크로 “ibr 140mg”라고 표시된 흰색 불투명 캡슐입니다.
정제:
140mg 정제는 한쪽 면에는 “ibr”가, 다른 쪽 면에는 “140”이 새겨진 황록색에서 녹색의 원형 정제입니다.
280mg 정제는 한쪽 면에는 “ibr”가, 다른 쪽 면에는 “280”이 새겨진 자주색 타원형 정제입니다.
420mg 정제는 한쪽 면에는 “ibr”가, 다른 쪽 면에는 “420”이 새겨진 황록색에서 녹색의 타원형 정제입니다.
경구 현탁액:
70mg/mL, 흰색에서 회백색 현탁액.
4 금기 사항
없음
5 경고 및 주의사항
5.1
출혈
IMBRUVICA를 투여받은 환자에서 치명적인 출혈 사건이 발생했습니다. 주요 출혈(≥ 3등급, 중증 또는 중추신경계 사건 포함; 예: 두개내 출혈[경막하혈종 포함], 위장관 출혈, 혈뇨 및 시술 후 출혈)은 27건의 임상시험에서 IMBRUVICA를 투여받은 2,838명의 환자 중 4.2%에서 발생했으며, 사망은 0.4%에서 발생했습니다. 멍과 점상출혈을 포함한 모든 등급의 출혈은 IMBRUVICA를 투여받은 환자의 각각 39%와 멍과 점상출혈을 제외하고 23%에서 발생했습니다 [유해 반응 (6.1)] 참조.
출혈 사건의 기전은 잘 알려져 있지 않습니다.
항응고제 또는 항혈소판제를 IMBRUVICA와 병용하면 주요 출혈 위험이 증가합니다. 임상시험 전반에 걸쳐 항혈소판제 또는 항응고제 치료 없이 IMBRUVICA를 투여받은 2,838명의 환자 중 3.1%에서 주요 출혈이 발생했습니다. 항혈소판제 치료를 항응고제 치료와 병용하거나 단독으로 사용하면 이 비율이 4.4%로 증가했으며, 항응고제 치료를 항혈소판제 치료와 병용하거나 단독으로 사용하면 이 비율이 6.1%로 증가했습니다. IMBRUVICA와 병용 투여할 때 항응고제 또는 항혈소판제 치료의 위험과 이점을 고려하십시오. 출혈의 징후와 증상을 모니터링하십시오.
수술 유형과 출혈 위험에 따라 수술 전후 최소 3일에서 7일 동안 IMBRUVICA 투여를 중단하는 것의 이점과 위험을 고려하십시오 [임상 연구 (14)] 참조.
5.2
감염
IMBRUVICA 치료와 함께 치명적인 감염과 치명적이지 않은 감염(세균성, 바이러스성 또는 곰팡이성 감염 포함)이 발생했습니다. 임상시험에서 IMBRUVICA를 투여받은 B 세포 악성 종양 환자 1,476명 중 21%에서 3등급 이상의 감염이 발생했습니다 [유해 반응 (6.1, 6.2)] 참조. 진행성 다초점 백질뇌병증(PML)과 폐렴구균 폐렴(PJP) 사례가 IMBRUVICA로 치료받은 환자에게서 발생했습니다. 기회 감염 위험이 높은 환자의 경우 표준 치료에 따라 예방 조치를 고려하십시오. 발열과 감염에 대해 환자를 모니터링하고 평가하고 적절하게 치료하십시오.
5.3
심장 부정맥, 심부전 및 돌연사
IMBRUVICA와 함께 치명적인 심각한 심장 부정맥과 심부전이 발생했습니다. 임상시험에서 IMBRUVICA를 투여받은 4,896명의 환자 중 1%에서 심장 질환 또는 돌연사로 인한 사망이 발생했으며, 여기에는 승인되지 않은 단독 요법 또는 병용 요법으로 IMBRUVICA를 투여받은 환자도 포함됩니다. 이러한 유해 반응은 기존 고혈압 또는 심혈관 질환이 있는 환자와 없는 환자 모두에서 발생했습니다. 심혈관 질환이 있는 환자는 이러한 사건의 위험이 더 클 수 있습니다.
3등급 이상의 심실 빈맥은 0.2%, 3등급 이상의 심방세동 및 심방 flutter는 3.7%, 3등급 이상의 심부전은 임상시험에서 IMBRUVICA를 투여받은 4,896명의 환자 중 1.3%에서 보고되었으며, 여기에는 승인되지 않은 단독 요법 또는 병용 요법으로 IMBRUVICA를 투여받은 환자도 포함됩니다. 이러한 사건은 특히 고혈압 및 당뇨병, 이전 심장 부정맥 병력이 있는 환자와 급성 감염이 있는 환자에서 발생했습니다 [유해 반응 (6.1)] 참조.
기준선에서 심장 병력 및 기능을 평가하고 심장 부정맥 및 심장 기능에 대해 환자를 모니터링하십시오. 부정맥 증상(예: 두근거림, 어지러움, 실신, 가슴 통증), 신규 발병 호흡 곤란 또는 기타 심혈관 문제가 발생하는 환자의 경우 필요에 따라 추가 평가(예: 심전도, 심초음파)를 실시하십시오. 심장 부정맥과 심부전을 적절하게 관리하고, 용량 수정 지침을 따르십시오 [용법 및 용량 (2.2)] 참조하고, IMBRUVICA 치료를 계속하는 것의 위험과 이점을 고려하십시오.
5.4
고혈압
임상시험에서 IMBRUVICA를 투여받은 B 세포 악성 종양 환자 1,476명 중 19%에서 고혈압이 발생했습니다. 환자의 8%에서 3등급 이상의 고혈압이 발생했습니다 [유해 반응 (6.1)] 참조. 이러한 환자의 하위 집단(N=1,124)의 데이터를 기반으로 중앙값 발병 시간은 5.9개월(범위, 0~24개월)이었습니다. B 세포 악성 종양 환자 1,284명에 대한 5년 이상의 장기 안전성 분석에서 중앙값 36개월(범위, 0~98개월) 동안 치료받은 환자의 고혈압 누적률은 시간이 지남에 따라 증가했습니다. 3등급 이상의 고혈압 유병률은 4%(0-1년), 7%(1-2년), 9%(2-3년), 9%(3-4년), 9%(4-5년)였으며, 5년 기간 동안 전체 발생률은 11%였습니다.
IMBRUVICA로 치료받는 환자의 혈압을 모니터링하고, IMBRUVICA 치료 전반에 걸쳐 적절하게 항고혈압제를 시작하거나 조정하고, 3등급 이상의 고혈압에 대한 용량 수정 지침을 따르십시오 [용법 및 용량 (2.2)] 참조.
5.5
혈구감소증
단일 요법으로 IMBRUVICA를 투여받은 B 세포 악성 종양 환자 645명에서, 실험실 측정 결과에 따르면 3등급 또는 4등급 호중구 감소증이 23%의 환자에게서 발생하였고, 3등급 또는 4등급 혈소판 감소증이 8%의 환자에게서, 그리고 3등급 또는 4등급 빈혈이 2.8%의 환자에게서 발생하였습니다 [유해 반응 (6.1)]을 참조하십시오.
매달 완전 혈구 수를 모니터링하십시오.
5.6
제2의 원발 악성 종양
임상 시험에서 IMBRUVICA를 투여받은 B 세포 악성 종양 환자 1,476명 중 비피부암종(3.9%)을 포함한 다른 악성 종양이 10%에서 발생했습니다 [유해 반응 (6.1)]을 참조하십시오. 가장 흔한 제2의 원발 악성 종양은 비흑색종 피부암(6%)이었습니다.
5.7
간독성(약물 유발 간 손상 포함)
IMBRUVICA를 포함한 Bruton 티로신 키나아제 억제제로 치료받은 환자에게서 중증, 생명을 위협하는, 그리고 잠재적으로 치명적인 약물 유발 간 손상(DILI)을 포함한 간독성이 발생했습니다.
기준선 및 IMBRUVICA 치료 전반에 걸쳐 빌리루빈과 트랜스아미나제를 평가하십시오. IMBRUVICA 투여 후 이상 간 기능 검사 결과가 나타나는 환자의 경우, 간 기능 검사 이상 및 간 독성의 임상 징후와 증상을 더 자주 모니터링하십시오. DILI가 의심되는 경우 IMBRUVICA 투여를 중단하십시오. DILI가 확인되면 IMBRUVICA 투여를 중단하십시오.
5.8
종양 용해 증후군
IMBRUVICA와 관련하여 종양 용해 증후군이 드물게 보고되었습니다 [유해 반응 (6.2)]을 참조하십시오. 기준 위험(예: 높은 종양 부담)을 평가하고 적절한 예방 조치를 취하십시오. 환자를 면밀히 모니터링하고 적절하게 치료하십시오.
5.9
배아-태아 독성
동물 연구 결과에 따르면, IMBRUVICA는 임산부에게 투여될 경우 태아에게 해를 끼칠 수 있습니다. 기관 형성기에 임신한 랫트와 토끼에게 이브루티닙을 투여한 결과, 혈액 악성 종양 환자에게서 보고된 수치보다 3~20배 높은 노출량에서 기형을 포함한 배아-태아 독성이 발생했습니다. 임신부에게 태아에 대한 잠재적 위험을 알리십시오. 임신 가능성이 있는 여성에게는 IMBRUVICA 치료 중 및 마지막 투여 후 1개월 동안 효과적인 피임법을 사용하도록 조언하십시오. [특정 환자군에서의 사용 (8.1)]을 참조하십시오.
6 부작용 반응
다음의 임상적으로 유의미한 이상반응은 라벨의 다른 곳에 설명되어 있습니다.
- 출혈 [경고 및 주의사항 (5.1) 참조]
- 감염 [경고 및 주의사항 (5.2) 참조]
- 심장 부정맥, 심부전 및 급사 [경고 및 주의사항 (5.3) 참조]
- 고혈압 [경고 및 주의사항 (5.4) 참조]
- 혈구감소증 [경고 및 주의사항 (5.5) 참조]
- 두 번째 원발성 악성종양 [경고 및 주의사항 (5.6) 참조]
- DILI를 포함한 간독성 [경고 및 주의사항 (5.7) 참조]
- 종양 용해 증후군 [경고 및 주의사항 (5.8) 참조]
6.1
임상시험 경험
임상시험은 매우 다양한 조건에서 수행되므로, 한 약물의 임상시험에서 관찰된 이상반응 발생률을 다른 약물의 임상시험 발생률과 직접 비교할 수 없으며 실제로 관찰된 발생률을 반영하지 않을 수 있습니다.
별도로 명시되지 않는 한, 경고 및 주의사항에 설명된 통합 안전성 모집단은 6건의 시험에서 IMBRUVICA에 대한 노출을 반영합니다. IMBRUVICA는 B세포 악성종양 환자에서 1일 1회 420mg 경구 단독 투여(475명), 1일 1회 560mg 경구 단독 투여[권장 성인 용량의 1.3배(174명)], 그리고 다른 약물과 병용하여 1일 1회 420mg 경구 투여(827명)되었습니다. 1,476명의 이 통합 안전성 모집단에서 87%는 6개월 이상 노출되었고 68%는 1년 이상 노출되었습니다. 가장 흔한 이상반응(≥ 30%)은 혈소판감소증, 설사, 피로, 근골격계 통증, 호중구감소증, 발진, 빈혈, 멍, 메스꺼움이었습니다.
경고 및 주의사항의 특정 하위 섹션에는 승인되지 않은 단독 요법 또는 병용 요법으로 IMBRUVICA를 투여받은 환자가 포함됩니다.
만성 림프구성 백혈병/소림프구성 림프종
아래에 설명된 데이터는 CLL/SLL 환자를 대상으로 한 단일군, 공개형 임상시험 1건(연구 1102) 및 무작위 대조 임상시험 5건(RESONATE, RESONATE-2, HELIOS, iLLUMINATE, E1912)에서 IMBRUVICA에 대한 노출을 반영합니다(총 n=2,016명, IMBRUVICA에 노출된 환자 n=1,133명 포함). 일반적으로 크레아티닌 청소율(CLcr) ≤ 30mL/min, AST 또는 ALT ≥ 2.5 x ULN, 또는 총 빌리루빈 ≥ 1.5 x ULN(비간성 원인 제외)인 환자는 이러한 시험에서 제외되었습니다. 연구 E1912에서는 AST 또는 ALT > 3 x ULN 또는 총 빌리루빈 > 2.5 x ULN인 환자가 제외되었습니다. 연구 1102에는 이전에 치료받은 CLL/SLL 환자 51명이 포함되었습니다. RESONATE에는 이전에 치료받은 CLL 또는 SLL 환자 386명이 무작위로 배정되어 단독 IMBRUVICA 또는 오파투무맙을 투여받았습니다. RESONATE-2에는 치료 경험이 없는 65세 이상의 CLL 또는 SLL 환자 267명이 무작위로 배정되어 단독 IMBRUVICA 또는 클로람부실을 투여받았습니다. HELIOS에는 이전에 치료받은 CLL 또는 SLL 환자 574명이 무작위로 배정되어 BR과 병용 IMBRUVICA 또는 위약과 병용 BR을 투여받았습니다. iLLUMINATE에는 치료 경험이 없는 65세 이상이거나 동반 질환이 있는 CLL/SLL 환자 228명이 무작위로 배정되어 오비누투주맙과 병용 IMBRUVICA 또는 오비누투주맙과 병용 클로람부실을 투여받았습니다. E1912에는 이전에 치료받지 않은 70세 이하의 CLL/SLL 환자 510명이 포함되었으며, 리툭시맙과 병용 IMBRUVICA 또는 플루다라빈, 시클로포스파미드, 리툭시맙(FCR)을 투여받았습니다.
IMBRUVICA를 투여받은 CLL/SLL 환자에서 가장 흔한 이상반응(≥ 30%)은 혈소판감소증, 설사, 피로, 근골격계 통증, 호중구감소증, 발진, 빈혈, 멍, 메스꺼움이었습니다.
IMBRUVICA를 투여받은 CLL/SLL 환자의 4~10%는 이상반응으로 인해 치료를 중단했습니다. 여기에는 폐렴, 출혈, 심방세동, 호중구감소증, 관절통, 발진, 혈소판감소증이 포함됩니다. 용량 감량으로 이어진 이상반응은 약 9%의 환자에서 발생했습니다.
연구 1102
이전에 치료받은 CLL/SLL 환자에게 1일 420mg의 단독 IMBRUVICA를 사용한 연구 1102(N=51)의 이상반응 및 실험실 이상은 중앙값 치료 기간이 15.6개월이며 발생률이 ≥ 10%인 경우 표 5 및 표 6에 제시되어 있습니다.
| 신체 기관계 | 이상반응 | 모든 등급 (%) | 3등급 이상 (%) |
| 위장관 장애 | 설사 변비 메스꺼움 구내염 구토 복통 소화불량 |
59 22 20 20 18 14 12 |
4 2 2 0 2 0 0 |
| 피부 및 피하 조직 장애 | 멍 발진 점상출혈 |
51 25 16 |
2 0 0 |
| 감염 및 감염증 | 상기도 감염 부비동염 피부 감염 폐렴 요로 감염 |
47 22 16 12 12 |
2 6 6 10 2 |
| 전신 장애 및 투여 부위 상태 | 피로 발열 말초 부종 무력증 오한 |
33 24 22 14 12 |
6 2 0 6 0 |
| 근골격계 및 결합 조직 장애 | 근골격계 통증 관절통 근육 경련 |
25 24 18 |
6 0 2 |
| 호흡기계, 흉부 및 종격 장애 | 기침 구인두 통증 호흡곤란 |
22 14 12 |
0 0 0 |
| 신경계 장애 | 현기증 두통 |
20 18 |
0 2 |
| 혈관 장애 | 고혈압 | 16 | 8 |
| 대사 및 영양 장애 | 식욕 감퇴 | 16 | 2 |
| 양성, 악성, 불특정 종양 | 이차 악성 종양 | 10 | 2† |
†조직구성 육종으로 인한 한 명의 환자 사망.
| 환자 비율 (N=51) | ||
| 모든 등급 (%) | 3등급 또는 4등급 (%) | |
| 혈소판 감소 | 69 | 12 |
| 호중구 감소 | 53 | 26 |
| 헤모글로빈 감소 | 43 | 0 |
* IWCLL 기준 및 이상반응에 따른 실험실 측정 결과 기반.
환자에서 치료 후 발생한 4등급 혈소판감소증(8%) 및 호중구감소증(12%)이 발생했습니다.
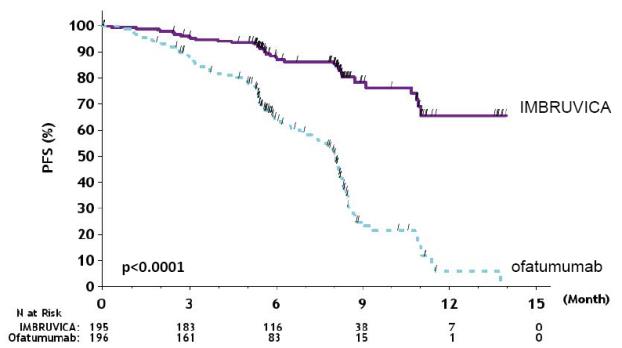
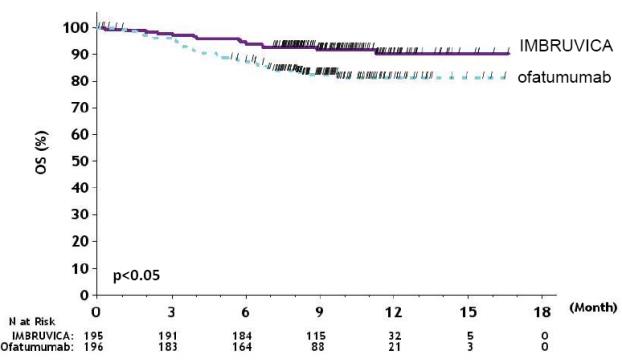
RESONATE
아래 표 7 및 표 8에 설명된 이상반응 및 실험실 이상은 이전에 치료받은 CLL/SLL 환자를 대상으로 한 RESONATE에서 IMBRUVICA에 대한 중앙값 8.6개월 노출 및 ofatumumab에 대한 중앙값 5.3개월 노출을 반영합니다.
| 신체기관계 이상반응 |
IMBRUVICA (N=195) |
Ofatumumab (N=191) |
||
| 모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
| 위장관계 장애 | ||||
| 설사 | 48 | 4 | 18 | 2 |
| 메스꺼움 | 26 | 2 | 18 | 0 |
| 구내염* | 17 | 1 | 6 | 1 |
| 변비 | 15 | 0 | 9 | 0 |
| 구토 | 14 | 0 | 6 | 1 |
| 근골격계 및 결합 조직 장애 | ||||
| 근골격계 통증* | 28 | 2 | 18 | 1 |
| 관절통 | 17 | 1 | 7 | 0 |
| 근육 경련 | 13 | 0 | 8 | 0 |
| 피부 및 피하 조직 장애 | ||||
| 발진* | 24 | 3 | 13 | 0 |
| 점상출혈 | 14 | 0 | 1 | 0 |
| 멍* | 12 | 0 | 1 | 0 |
| 전신 장애 및 투여 부위 상태 | ||||
| 발열 | 24 | 2 | 15 | 2† |
| 호흡기계, 흉부 및 종격 장애 | ||||
| 기침 | 19 | 0 | 23 | 1 |
| 호흡곤란 | 12 | 2 | 10 | 1 |
| 감염 및 감염성 질환 | ||||
| 상기도 감염 | 16 | 1 | 11 | 2† |
| Pneumonia* | 15 | 12† | 13 | 10† |
| Sinusitis* | 11 | 1 | 6 | 0 |
| 요로 감염 | 10 | 4 | 5 | 1 |
| 신경계 장애 | ||||
| 두통 | 14 | 1 | 6 | 0 |
| 현기증 | 11 | 0 | 5 | 0 |
| 손상, 중독 및 시술 합병증 | ||||
| 타박상 | 11 | 0 | 3 | 0 |
| 눈 장애 | ||||
| 시야 흐림 | 10 | 0 | 3 | 0 |
| 신체 기관계 및 개별 ADR 용어는 IMBRUVICA군에서 발생 빈도의 내림차순으로 정렬되어 있습니다. * 여러 ADR 용어를 포함합니다. † 각 군에서 사망으로 이어진 폐렴 3건과 ofatumumab군에서 사망으로 이어진 발열 및 상기도 감염 1건을 포함합니다. |
||||
| IMBRUVICA (N=195) |
Ofatumumab (N=191) |
|||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 호중구 감소 | 51 | 23 | 57 | 26 |
| 혈소판 감소 | 52 | 5 | 45 | 10 |
| 헤모글로빈 감소 | 36 | 0 | 21 | 0 |
치료로 발생한 4등급 혈소판감소증(IMBRUVICA군 2% vs ofatumumab군 3%) 및 호중구감소증(IMBRUVICA군 8% vs ofatumumab군 8%)이 환자에서 발생했습니다.
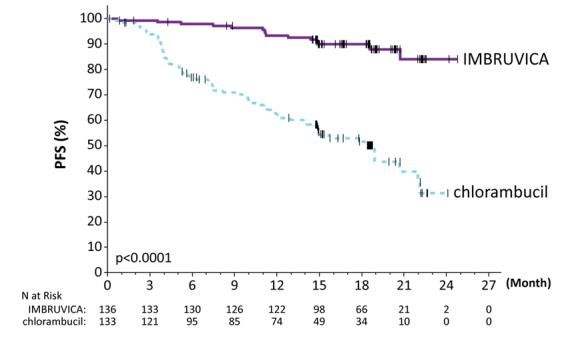
RESONATE-2
아래 표 9 및 표 10에 설명된 이상반응 및 실험실 이상은 IMBRUVICA에 대한 중앙값 17.4개월의 노출을 반영합니다. RESONATE-2에서 chlorambucil에 대한 중앙값 노출은 7.1개월이었습니다.
| 신체 기관계 이상반응 |
IMBRUVICA (N=135) |
Chlorambucil (N=132) |
||
| 모든 등급 (%) |
3등급 이상 (%) | 모든 등급 (%) |
3등급 이상 (%) | |
| 위장관계 장애 | ||||
| 설사 | 42 | 4 | 17 | 0 |
| 메스꺼움 | 22 | 1 | 39 | 1 |
| 변비 | 16 | 1 | 16 | 0 |
| 구내염* | 14 | 1 | 4 | 1 |
| 구토 | 13 | 0 | 20 | 1 |
| 복통 | 13 | 3 | 11 | 1 |
| 소화불량 | 11 | 0 | 2 | 0 |
| 근골격계 및 결합 조직 장애 | ||||
| 근골격계 통증* | 36 | 4 | 20 | 0 |
| 관절통 | 16 | 1 | 7 | 1 |
| 근육 경련 | 11 | 0 | 5 | 0 |
| 전신 장애 및 투여 부위 상태 | ||||
| 피로 | 30 | 1 | 38 | 5 |
| 말초 부종 | 19 | 1 | 9 | 0 |
| 발열 | 17 | 0 | 14 | 2 |
| 호흡기계, 흉부 및 종격동 장애 | ||||
| 기침 | 22 | 0 | 15 | 0 |
| 호흡곤란 | 10 | 1 | 10 | 0 |
| 피부 및 피하 조직 장애 | ||||
| 발진* | 21 | 4 | 12 | 2 |
| 멍* | 19 | 0 | 7 | 0 |
| 눈 장애 | ||||
| 안구건조증 | 17 | 0 | 5 | 0 |
| 눈물흘림 증가 | 13 | 0 | 6 | 0 |
| 시야 흐림 | 13 | 0 | 8 | 0 |
| 시력 감소 | 11 | 0 | 2 | 0 |
| 감염 및 감염증 | ||||
| 상기도 감염 | 17 | 2 | 17 | 2 |
| 피부 감염* | 15 | 2 | 3 | 1 |
| 폐렴* | 14 | 8 | 7 | 4 |
| 요로 감염 | 10 | 1 | 8 | 1 |
| 혈관 장애 | ||||
| 고혈압* | 14 | 4 | 1 | 0 |
| 신경계 장애 | ||||
| 두통 | 12 | 1 | 10 | 2 |
| 현기증 | 11 | 0 | 12 | 1 |
| 검사 | ||||
| 체중 감소 | 10 | 0 | 12 | 0 |
주어진 ADR 용어에 대해 여러 이벤트가 있는 대상은 각 ADR 용어에 대해 한 번만 계산됩니다.
신체 시스템 및 개별 ADR 용어는 IMBRUVICA군에서 발생 빈도의 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
| IMBRUVICA (N=135) |
Chlorambucil (N=132) |
|||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| Neutrophils Decreased | 55 | 28 | 67 | 31 |
| Platelets Decreased | 47 | 7 | 58 | 14 |
| Hemoglobin Decreased | 36 | 0 | 39 | 2 |
치료로 발생한 4등급 혈소판감소증(IMBRUVICA군 1% vs chlorambucil군 3%)과 호중구감소증(IMBRUVICA군 11% vs chlorambucil군 12%)이 환자에서 발생했습니다.
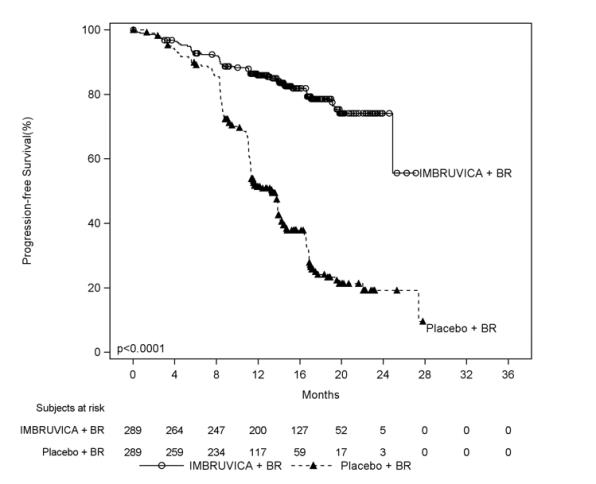
HELIOS
아래 표 11에 설명된 이상반응은 이전에 치료받은 CLL/SLL 환자를 대상으로 한 HELIOS 연구에서 IMBRUVICA + BR에 대한 중앙값 14.7개월 노출과 위약 + BR에 대한 중앙값 12.8개월 노출을 반영합니다.
| 신체 기관 이상반응 |
IMBRUVICA + BR (N=287) |
위약 + BR (N=287) |
||
| 모든 등급 (%) |
3등급 이상 (%) | 모든 등급 (%) |
3등급 이상 (%) | |
| 혈액 및 림프계 장애 | ||||
| Neutropenia* | 66 | 61 | 60 | 56† |
| Thrombocytopenia* | 34 | 16 | 26 | 16 |
| 위장관계 장애 | ||||
| Diarrhea | 36 | 2 | 23 | 1 |
| Abdominal pain | 12 | 1 | 8 | <1 |
| 피부 및 피하 조직 장애 | ||||
| Rash* | 32 | 4 | 25 | 1 |
| Bruising * | 20 | <1 | 8 | <1 |
| 근골격계 및 결합 조직 장애 | ||||
| Musculoskeletal pain* | 29 | 2 | 20 | 0 |
| Muscle spasms | 12 | <1 | 5 | 0 |
| 전신 장애 및 투여 부위 상태 | ||||
| Pyrexia | 25 | 4 | 22 | 2 |
| 혈관 장애 | ||||
| Hemorrhage* | 19 | 2† | 9 | 1 |
| 고혈압* | 11 | 5 | 5 | 2 |
| 감염 및 감염증 | ||||
| 기관지염 | 13 | 2 | 10 | 3 |
| 피부 감염* | 10 | 3 | 6 | 2 |
| 대사 및 영양 장애 | ||||
| 고요산혈증 | 10 | 2 | 6 | 0 |
IMBRUVICA군에서 신체 기관계 및 개별 ADR 용어는 빈도 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
<1은 0보다 크고 0.5% 미만인 빈도에 사용됩니다.
† IMBRUVICA군에서 치명적인 결과를 초래한 출혈 사례 2건과 위약 + BR군에서 치명적인 결과를 초래한 호중구 감소증 사례 1건이 포함됩니다.
모든 등급의 심방세동은 IMBRUVICA + BR로 치료받은 환자의 7%와 위약 + BR로 치료받은 환자의 2%에서 발생했습니다. 3등급 및 4등급 심방세동의 빈도는 IMBRUVICA + BR로 치료받은 환자의 3%와 위약 + BR로 치료받은 환자의 1%였습니다.
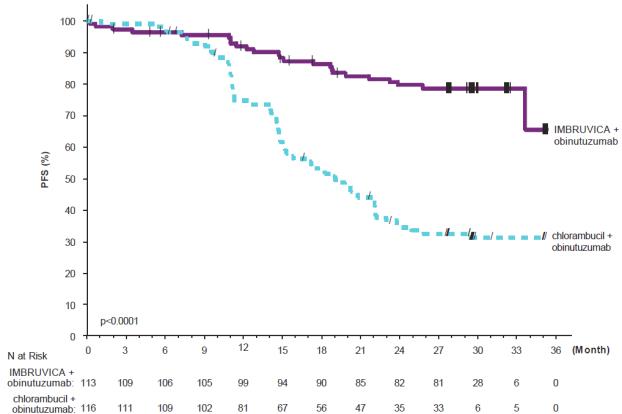
iLLUMINATE
아래 표 12에 설명된 이상반응은 이전에 치료받지 않은 CLL/SLL 환자를 대상으로 한 iLLUMINATE에서 IMBRUVICA + obinutuzumab에 대한 중앙값 29.3개월의 노출과 chlorambucil + obinutuzumab에 대한 중앙값 5.1개월의 노출을 반영합니다.
| 신체 기관계 이상반응 |
IMBRUVICA + Obinutuzumab (N=113) |
Chlorambucil + Obinutuzumab (N=115) |
||
| 모든 등급 (%) |
3등급 이상 (%) | 모든 등급 (%) |
3등급 이상 (%) | |
| 혈액 및 림프계 장애 | ||||
| Neutropenia* | 48 | 39 | 64 | 48 |
| Thrombocytopenia* | 36 | 19 | 28 | 11 |
| Anemia | 17 | 4 | 25 | 8 |
| 피부 및 피하 조직 장애 | ||||
| Rash* | 36 | 3 | 11 | 0 |
| Bruising* | 32 | 3 | 3 | 0 |
| 위장관 장애 | ||||
| Diarrhea | 34 | 3 | 10 | 0 |
| Constipation | 16 | 0 | 12 | 1 |
| Nausea | 12 | 0 | 30 | 0 |
| 근골격계 및 결합 조직 장애 | ||||
| Musculoskeletal pain* | 33 | 1 | 23 | 3 |
| Arthralgia | 22 | 1 | 10 | 0 |
| Muscle spasms | 13 | 0 | 6 | 0 |
| 호흡기계, 흉부 및 종격 질환 | ||||
| 기침 | 27 | 1 | 12 | 0 |
| 손상, 중독 및 시술 합병증 | ||||
| Infusion related reaction | 25 | 2 | 58 | 8 |
| 혈관 질환 | ||||
| Hemorrhage* | 25 | 1 | 9 | 0 |
| 고혈압* | 17 | 4 | 4 | 3 |
| 전신 질환 및 투여 부위 상태 | ||||
| 발열 | 19 | 2 | 26 | 1 |
| 피로 | 18 | 0 | 17 | 2 |
| Peripheral edema | 12 | 0 | 7 | 0 |
| 감염 및 감염성 질환 | ||||
| Pneumonia* | 16 | 9 | 9 | 4† |
| 상기도 감염 | 14 | 1 | 6 | 0 |
| Skin infection* | 13 | 1 | 3 | 0 |
| 요로 감염 | 12 | 3 | 7 | 1 |
| Nasopharyngitis | 12 | 0 | 3 | 0 |
| 결막염 | 11 | 0 | 2 | 0 |
| 대사 및 영양 장애 | ||||
| Hyperuricemia | 13 | 1 | 0 | 0 |
| 심장 질환 | ||||
| Atrial fibrillation | 12 | 5 | 0 | 0 |
| 정신 질환 | ||||
| 불면증(Insomnia) | 12 | 0 | 4 | 0 |
IMBRUVICA군에서 신체 기관계 및 개별 ADR 용어는 빈도 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
† 사망으로 이어진 한 가지 사례를 포함합니다.
E1912
아래 표 13에 설명된 이상반응은 이전에 치료받지 않은 70세 이하의 CLL/SLL 환자를 대상으로 한 E1912에서 IMBRUVICA + rituximab에 대한 중앙값 34.3개월의 노출과 FCR에 대한 중앙값 4.7개월의 노출을 반영합니다.
| 신체 기관계 이상반응 |
IMBRUVICA + Rituximab
(N=352) |
Fludarabine + Cyclophosphamide + Rituximab (N=158) |
||
| 모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
| 전신 장애 및 투여 부위 이상 | ||||
| 피로 | 80 | 2 | 78 | 3 |
| 말초 부종 | 28 | 1 | 17 | 0 |
| 발열 | 27 | 1 | 27 | 1 |
| 통증 | 23 | 2 | 8 | 0 |
| 근골격계 및 결합 조직 장애 | ||||
| 근골격계 통증* | 61 | 5 | 35 | 2 |
| 관절통 | 41 | 5 | 10 | 1 |
| 위장관 장애 | ||||
| 설사 | 53 | 4 | 27 | 1 |
| 메스꺼움 | 40 | 1 | 64 | 1 |
| 구내염* | 22 | 1 | 8 | 1 |
| 복통* | 19 | 2 | 10 | 1 |
| 구토 | 18 | 2 | 28 | 0 |
| 변비 | 17 | 0 | 32 | 0 |
| 피부 및 피하 조직 장애 | ||||
| 발진* | 49 | 4 | 29 | 5 |
| 멍* | 36 | 1 | 4 | 1 |
| Vascular disorders | ||||
| 고혈압* | 42 | 19 | 22 | 6 |
| 출혈* | 31 | 2 | 8 | 1 |
| Nervous system disorders | ||||
| 두통 | 40 | 1 | 27 | 1 |
| 현기증 | 21 | 1 | 13 | 1 |
| Peripheral neuropathy* | 19 | 1 | 13 | 1 |
| Respiratory, thoracic and mediastinal disorders | ||||
| 기침 | 32 | 0 | 25 | 0 |
| 호흡곤란 | 22 | 2 | 21 | 1 |
| Infections and infestations | ||||
| 상기도 | 29 | 1 | 19 | 2 |
| 감염 | ||||
| 피부 감염* | 16 | 1 | 3 | 1 |
| Metabolism and nutrition disorders | ||||
| Hyperuricemia | 19 | 1 | 4 | 0 |
| 식욕 감퇴 | 15 | 0 | 20 | 1 |
| Psychiatric disorders | ||||
| 불면증 | 16 | 1 | 19 | 1 |
IMBRUVICA군에서 신체 기관계 및 개별 ADR 용어는 빈도 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
| IMBRUVICA + Rituximab
(N=352) |
Fludarabine + Cyclophosphamide + Rituximab (N=158) |
|||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 혈액학적 이상 호중구 감소 혈소판 감소 헤모글로빈 감소 |
53 43 26 |
30 7 0 |
70 69 51 |
44 25 2 |
| 화학적 이상 크레아티닌 증가 빌리루빈 증가 AST 증가 |
38 30 25 |
1 2 3 |
17 15 23 |
1 0 <1 |
IWCLL 기준에 따른 실험실 측정 기반.
월든스트롬 마크로글로불린혈증
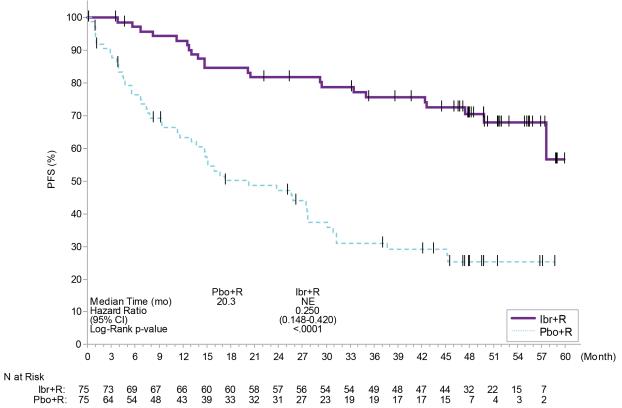
아래에 설명된 데이터는 두 건의 단일군 임상 시험(1118 연구 및 INNOVATE 단독 요법군)과 한 건의 무작위 대조 시험(INNOVATE)에서 IMBRUVICA에 노출된 총 169명의 WM 환자를 포함하여 IMBRUVICA에 대한 노출을 반영합니다. 1118 연구에는 이전에 치료받은 WM 환자 63명이 포함되었으며, 이들은 단일 제제 IMBRUVICA를 투여받았습니다. INNOVATE에는 치료 경험이 없거나 이전에 치료받은 WM 환자 150명이 포함되었으며, 이들은 리툭시맙과 병용하여 IMBRUVICA 또는 위약을 투여받았습니다. INNOVATE 단독 요법군에는 이전 리툭시맙 병용 요법 실패 후 IMBRUVICA를 투여받은 이전에 치료받은 WM 환자 31명이 포함되었습니다.
1118 연구 및 INNOVATE(≥ 20%)에서 가장 흔한 이상반응은 호중구 감소증, 설사, 멍, 혈소판 감소증, 출혈, 근골격계 통증, 발진 및 메스꺼움이었습니다.
1118 연구 및 INNOVATE에서 IMBRUVICA를 투여받은 환자의 5%가 이상반응으로 인해 치료를 중단했습니다. 치료 중단으로 이어진 가장 흔한 이상반응은 심방세동이었습니다. 용량 감량으로 이어진 이상반응은 환자의 14%에서 발생했습니다.
1118 연구 및 INNOVATE 단독 요법군
표 15 및 표 16에 설명된 이상반응 및 실험실 이상은 1118 연구에서 11.7개월, INNOVATE 단독 요법군에서 33개월의 중앙값 기간 동안 IMBRUVICA에 대한 노출을 반영합니다.
| 신체 기관 | 이상반응 | 모든 등급 (%) | 3등급 이상 (%) |
| 위장관 장애 | 설사 메스꺼움 구내염* 변비 위식도 역류 질환 |
38 21 15 12 12 |
2 0 0 1 0 |
| 피부 및 피하 조직 장애 | 멍* 발진* |
28 21 |
1 1 |
| 혈관 장애 | 출혈* 고혈압* |
28 14 |
0 4 |
| 전신 장애 및 투여 부위 상태 | 피로 발열 |
18 12 |
2 2 |
| 근골격계 및 결합 조직 장애 | 근골격계 통증* 근육 경련 |
21 19 |
0 0 |
| 감염 및 감염증 | 상기도 감염 피부 감염* 부비동염* 폐렴* |
19 18 16 13 |
0 3 0 5 |
| 신경계 장애 | 두통 현기증 |
14 13 |
0 0 |
| 호흡기계, 흉부 및 종격 장애 | 기침 | 13 | 0 |
신체 기관 및 개별 ADR 선호 용어는 발생 빈도의 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
| 환자 비율 (N=94) | ||
| 모든 등급 (%) | 3등급 또는 4등급 (%) | |
| 혈소판 감소 | 38 | 11 |
| 호중구 감소 | 43 | 16 |
| 헤모글로빈 감소 | 21 | 6 |
4등급 혈소판감소증(4%) 및 호중구감소증(7%)이 환자에서 발생했습니다.
INNOVATE
아래 표 17에 설명된 이상반응은 INNOVATE 연구에서 치료 경험이 없는 또는 이전에 치료받은 WM 환자에서 IMBRUVICA + R에 대한 중앙값 25.8개월 노출 및 위약 + R에 대한 중앙값 15.5개월 노출을 반영합니다.
| 신체기관 이상반응 |
IMBRUVICA + R (N=75) |
위약 + R (N=75) |
||
| 모든 등급 (%) |
3등급 이상 (%) |
모든 등급 (%) |
3등급 이상 (%) |
|
| 피부 및 피하 조직 장애 | ||||
| 멍* | 37 | 1 | 5 | 0 |
| 발진* | 24 | 1 | 11 | 0 |
| 근골격계 및 결합 조직 장애 | ||||
| 근골격계 통증* | 35 | 4 | 21 | 3 |
| 관절통 | 24 | 3 | 11 | 1 |
| 근육 경련 | 17 | 0 | 12 | 1 |
| 혈관 장애 | ||||
| 출혈* | 32 | 3 | 17 | 4† |
| 고혈압* | 20 | 13 | 5 | 4 |
| 위장관 장애 | ||||
| 설사 | 28 | 0 | 15 | 1 |
| 메스꺼움 | 21 | 0 | 12 | 0 |
| 소화불량 | 16 | 0 | 1 | 0 |
| 변비 | 13 | 1 | 11 | 1 |
| 감염 및 감염증 | ||||
| 폐렴* | 19 | 13 | 5 | 3 |
| 피부 감염* | 17 | 3 | 3 | 0 |
| 요로 감염 | 13 | 0 | 0 | 0 |
| 기관지염 | 12 | 3 | 7 | 0 |
| 인플루엔자 | 12 | 0 | 7 | 1 |
| 바이러스성 상기도 감염 | 11 | 0 | 7 | 0 |
| 전신 장애 및 투여 부위 이상 | ||||
| 말초 부종 | 17 | 0 | 12 | 1 |
| 호흡기, 흉부 및 종격 장애 | ||||
| 기침 | 17 | 0 | 11 | 0 |
| 혈액 및 림프계 장애 | ||||
| 호중구감소증* | 16 | 12 | 11 | 4 |
| 심장 장애 | ||||
| 심방세동 | 15 | 12 | 3 | 1 |
| 신경계 장애 | ||||
| 현기증 | 11 | 0 | 7 | 0 |
| 정신 장애 | ||||
| 불면증 | 11 | 0 | 4 | 0 |
| 대사 및 영양 장애 | ||||
| 저칼륨혈증 | 11 | 0 | 1 | 1 |
전신 및 개별 ADR 선호 용어는 발생 빈도의 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
† 사망으로 이어진 한 가지 사례를 포함합니다.
IR로 치료받은 환자의 1%에서 3등급 또는 4등급 주입 관련 반응이 관찰되었습니다.
만성 이식편대숙주병
연구 1129
아래에 설명된 데이터는 1차 코르티코스테로이드 치료 실패 후 cGVHD가 발생하고 추가 치료가 필요한 42명의 환자를 포함하는 공개형 임상 시험(연구 1129)에서 IMBRUVICA에 대한 노출을 반영합니다[임상 연구 (14.3)].
연구 1129에서 가장 흔한 이상반응(≥ 20%)은 피로, 멍, 설사, 혈소판 감소증, 구내염, 근육 경련, 메스꺼움, 출혈, 빈혈 및 폐렴이었습니다. 심방 세동은 한 환자(2%)에서 발생했으며 3등급이었습니다.
연구 1129에서 IMBRUVICA를 투여받은 환자의 24%가 이상반응으로 인해 치료를 중단했습니다. 치료 중단으로 이어진 가장 흔한 이상반응은 피로와 폐렴이었습니다. 용량 감소로 이어진 이상반응은 환자의 26%에서 발생했습니다.
아래 표 18 및 표 19에 설명된 이상반응 및 실험실 이상은 연구 1129에서 IMBRUVICA에 대한 중앙값 4.4개월의 노출을 반영합니다.
| 신체 기관 | 이상반응 | 모든 등급 (%) | 3등급 이상 (%) |
| 전신 장애 및 투여 부위 이상 | 피로 발열 말초 부종 |
57 17 12 |
12 5 0 |
| 피부 및 피하 조직 장애 | 멍* 발진* |
40 12 |
0 0 |
| 위장관 장애 | 설사 구내염* 메스꺼움 변비 |
36 29 26 12 |
10 2 0 0 |
| 근골격계 및 결합 조직 장애 | 근육 경련 근골격계 통증* |
29 14 |
2 5 |
| 혈관 장애 | 출혈* | 26 | 0 |
| 감염 및 감염증 | 폐렴* 상기도 감염 패혈증* |
21 19 10 |
14† 0 10 |
| 신경계 장애 | 두통 | 17 | 5 |
| 손상, 중독 및 시술 합병증 | 낙상 | 17 | 0 |
| 호흡기, 흉부 및 종격 장애 | 기침 호흡곤란 |
14 12 |
0 2 |
| 대사 및 영양 장애 | 저칼륨혈증 | 12 | 7 |
시스템 장기 분류 및 개별 ADR 선호 용어는 발생 빈도의 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
† 사망으로 이어진 2건의 사례를 포함합니다.
| 환자 비율 (N=42) | ||
| 모든 등급 (%) | 3등급 또는 4등급 (%) | |
| Platelets decreased | 33 | 0 |
| Neutrophils decreased | 10 | 10 |
| Hemoglobin decreased | 24 | 2 |
4등급의 치료 발생 호중구 감소증이 환자의 2%에서 발생했습니다.
iMAGINE
IMBRUVICA의 안전성은 하나 이상의 전신 요법 실패 후 cGVHD가 있는 1세에서 22세 미만의 소아 및 청소년 환자 47명을 포함한 iMAGINE 연구에서 평가되었습니다. 12세 이상의 환자는 IMBRUVICA 420mg을 1일 1회 경구 투여했으며, 1세에서 12세 미만의 환자는 IMBRUVICA 240mg/m2을 1일 1회 경구 투여했습니다. [see Clinical Studies (14.3)]. IMBRUVICA에 대한 노출 기간 중앙값은 7.1개월(범위, 0.2~25.9개월)이었습니다.
IMBRUVICA를 투여받은 환자의 64%에서 심각한 이상반응이 발생했습니다. 두 명 이상의 환자에서 발생한 심각한 이상반응에는 폐렴, 발열, 패혈증, 구내염이 포함되었습니다. 패혈증 및 급성 호흡곤란 증후군(ARDS)을 포함하여 IMBRUVICA를 투여받은 두 명의 환자에서 치명적인 이상반응이 발생했습니다.
이상반응으로 인해 IMBRUVICA 투여를 영구적으로 중단한 환자는 23%였습니다. 두 명 이상의 환자에서 영구적인 투여 중단을 초래한 이상반응에는 출혈이 포함되었습니다. 이상반응으로 인해 IMBRUVICA의 용량을 감량한 환자는 19%였습니다. 두 명 이상의 환자에서 용량 감량이 필요한 이상반응에는 구내염이 포함되었습니다.
검사 이상을 포함하여 가장 흔한(≥ 20%) 이상반응은 빈혈, 근골격계 통증, 발열, 설사, 폐렴, 복통, 구내염, 혈소판 감소증, 두통이었습니다.
표 20에는 iMAGINE에서 발생한 이상반응이 요약되어 있습니다.
| IMBRUVICA (N=47) |
||
| 신체 기관 이상반응 |
모든 등급 (%) |
3등급 또는 4등급 (%) |
| 전신 장애 및 투여 부위 이상 | ||
| 발열 | 30 | 11 |
| 근골격계 및 결합 조직 장애 | ||
| 근골격계 통증* | 30 | 2 |
| 골괴사 | 11 | 9 |
| 위장관 장애 | ||
| 설사 | 28 | 2 |
| 복통* | 23 | 4 |
| 구내염* | 23 | 9 |
| 구토 | 19 | 2 |
| 메스꺼움 | 19 | 4 |
| 감염 및 감염성 질환 | ||
| 폐렴* | 23 | 13 |
| 피부 감염* | 17 | 4 |
| 패혈증* | 11 | 9† |
| 신경계 장애 | ||
| 두통 | 21 | 2 |
| 피부 및 피하 조직 장애 | ||
| 발진* | 19 | 2 |
| 가려움증 | 13 | 0 |
| 점상출혈 | 13 | 0 |
| 호흡기계, 흉부 및 종격 질환 | ||
| 기침 | 19 | 2 |
| 혈관 질환 | ||
| Hemorrhage* | 17 | 0 |
| 고혈압* | 11 | 4 |
| 혈액 및 림프계 질환 | ||
| 저칼륨혈증 | 15 | 6 |
| Hypogammaglobulinemia* | 11 | 0 |
| 심장 질환 | ||
| Sinus tachycardia | 11 | 0 |
| 검사 | ||
| Alanine aminotransferase increased | 11 | 2 |
전신 장기 등급 및 개별 ADR 선호 용어는 발생 빈도의 내림차순으로 정렬됩니다.
* 여러 ADR 용어를 포함합니다.
† 사망 1건을 포함합니다.
표 21 은 iMAGINE에서의 실험실 이상을 요약한 것입니다.
| IMBRUVICA (N=47) |
||
| 모든 등급 (%) |
3등급 또는 4등급 (%) |
|
| 헤모글로빈 감소 | 49 | 13 |
| 혈소판 감소 | 21 | 4 |
| 호중구 감소 | 13 | 6 |
4등급 치료 후 발생한 호중구 감소증은 환자의 3%에서 발생했습니다.
추가적인 중요 이상반응
심혈관계 사건
심혈관계 사건에 대한 데이터는 IMBRUVICA를 사용한 무작위 대조 시험(n=2,115; IMBRUVICA로 치료받은 1,157명의 환자의 경우 중앙값 치료 기간 19.1개월, 대조군의 958명의 환자의 경우 5.3개월)을 기반으로 합니다. 모든 등급의 심실 빈맥(심실 조기 수축, 심실 부정맥, 심실 세동, 심실 조동 및 심실 빈맥) 발생률은 IMBRUVICA로 치료받은 환자에서 대조군 환자에 비해 1.0% 대 0.4%였으며, 3등급 이상은 0.3% 대 0%였습니다. 모든 등급의 심방 세동 및 심방 조동 발생률은 IMBRUVICA로 치료받은 환자에서 대조군 환자에 비해 8.4% 대 1.6%였으며, 3등급 이상은 4.0% 대 0.5%였습니다. 또한, 모든 등급의 심부전 발생률은 IMBRUVICA로 치료받은 환자에서 대조군 환자에 비해 1.7% 대 0.5%였으며, 3등급 이상은 1.2% 대 0.3%였습니다.
모든 등급의 허혈성 뇌혈관 사건(뇌혈관 사고, 허혈성 뇌졸중, 뇌 허혈 및 일과성 허혈 발작) 발생률은 IMBRUVICA로 치료받은 환자에서 대조군 환자에 비해 각각 1% 대 0.4%였으며, 3등급 이상은 0.5% 대 0.2%였습니다.
설사
무작위 대조 시험(n=2,115; IMBRUVICA로 치료받은 1,157명의 환자의 경우 중앙값 치료 기간 19.1개월, 대조군의 958명의 환자의 경우 5.3개월)에서 모든 등급의 설사는 IMBRUVICA로 치료받은 환자의 43%에서 발생했으며, 대조군 환자의 경우 19%였습니다. 3등급 설사는 IMBRUVICA로 치료받은 환자의 3%에서 발생했으며, 대조군 환자의 경우 1%였습니다. 설사로 인해 IMBRUVICA 치료를 중단한 환자는 1% 미만(0.3%)이었으며, 대조군에서는 0%였습니다.
이 환자들 중 1,605명의 데이터를 기반으로, 모든 등급의 설사에 대한 첫 발생까지의 중앙값 시간은 IMBRUVICA로 치료받은 환자에서 21일(범위, 0~708일)이었으며, 대조군에서는 46일(범위, 0~492일)이었습니다. 3등급 설사의 경우 IMBRUVICA로 치료받은 환자에서 117일(범위, 3~414일)이었으며, 대조군에서는 194일(범위, 11~325일)이었습니다. 설사를 보고한 환자 중 완전히 해결된 환자는 IMBRUVICA로 치료받은 환자에서 85%, 대조군에서 89%였으며, 분석 시점에 해결되지 않은 환자는 각각 15%와 11%였습니다. IMBRUVICA로 치료받은 환자에서 발생에서 해결까지의 중앙값 시간은 모든 등급의 설사의 경우 7일(범위, 1~655일), 3등급 설사의 경우 7일(범위, 1~78일)이었으며, 대조군에서는 각각 4일(범위, 1~367일)과 19일(범위, 1~56일)이었습니다.
시각 장애
무작위 대조 시험(n=2,115; IMBRUVICA로 치료받은 1,157명의 환자의 경우 중앙값 치료 기간 19.1개월, 대조군의 958명의 환자의 경우 5.3개월)에서 모든 등급의 시야 흐림 및 시력 감소는 IMBRUVICA로 치료받은 환자의 11%(1등급 9%, 2등급 2%, 3등급 이상 없음)에서 발생했으며, 대조군에서는 6%(1등급 5%, 2등급 및 3등급 < 1%)였습니다.
이 환자들 중 1,605명의 데이터를 기반으로, 첫 발생까지의 중앙값 시간은 IMBRUVICA로 치료받은 환자에서 91일(범위, 0~617일)이었으며, 대조군에서는 100일(범위, 2~477일)이었습니다. 시각 장애를 보고한 환자 중 완전히 해결된 환자는 IMBRUVICA로 치료받은 환자에서 60%, 대조군에서 71%였으며, 분석 시점에 해결되지 않은 환자는 각각 40%와 29%였습니다. 발생에서 해결까지의 중앙값 시간은 IMBRUVICA로 치료받은 환자에서 37일(범위, 1~457일)이었으며, 대조군에서는 26일(범위, 1~721일)이었습니다.
6.2
Postmarketing Experience
IMBRUVICA의 시판 후 사용 중 다음과 같은 이상반응이 확인되었습니다. 이러한 반응은 불확실한 규모의 모집단에서 자발적으로 보고되기 때문에 항상 빈도를 확실하게 추정하거나 약물 노출과의 인과 관계를 확립할 수 있는 것은 아닙니다.
- 간담도 질환: 급성 및/또는 치명적인 사건을 포함한 간부전, 간경변, 약물 유발 간 손상
- 호흡기 질환: 간질성 폐 질환
- 대사 및 영양 장애: 종양 용해 증후군
- 면역 체계 장애: 아나필락시스 쇼크, 혈관 부종, 두드러기
- 피부 및 피하 조직 장애: Stevens-Johnson 증후군(SJS), 조갑박리증, 지방층염, 호중구성 피부병, 피부 혈관염
- 감염: B형 간염 재활성화
- 신경계 장애: 말초 신경병증
7 약물 상호 작용
7.1
CYP3A 저해제가 이브루티닙에 미치는 영향
IMBRUVICA와 강력하거나 중등도의 CYP3A 저해제를 병용 투여하면 이브루티닙 혈장 농도가 증가할 수 있습니다 [임상 약리학 (12.3)]를 참조하십시오. 이브루티닙 농도 증가는 약물 관련 독성 위험을 증가시킬 수 있습니다.
포사코나졸, 보리코나졸 및 중등도 CYP3A 저해제와 병용 투여하는 경우 IMBRUVICA의 용량 조절을 권장합니다 [용법 및 용량 (2.3)]를 참조하십시오.
다른 강력한 CYP3A 저해제와의 병용 투여는 피하십시오. 이러한 저해제를 단기간(예: 7일 이내의 항감염제) 사용할 경우 IMBRUVICA 투여를 중단하십시오 [용법 및 용량 (2.3)]를 참조하십시오.
IMBRUVICA 치료 중에는 자몽과 세비야 오렌지를 섭취하지 마십시오. 이러한 과일에는 강력하거나 중등도의 CYP3A 저해제가 함유되어 있습니다.
7.2
CYP3A 유도제가 이브루티닙에 미치는 영향
IMBRUVICA와 강력한 CYP3A 유도제를 병용 투여하면 이브루티닙 농도가 감소할 수 있습니다. 강력한 CYP3A 유도제와의 병용 투여는 피하십시오 [임상 약리학 (12.3)]를 참조하십시오.
8 특정 집단에서의 사용
8.1
임신
위험 요약
동물 연구 결과에 따르면 IMBRUVICA는 태아에게 해를 끼칠 수 있습니다. 임신 여성에 대한 IMBRUVICA 사용에 대한 데이터가 없으므로 주요 선천적 기형 및 유산의 약물 관련 위험을 알 수 없습니다. 동물 생식 연구에서 기관 형성 기간 동안 임신 랫트와 토끼에 대해 1일 420mg의 임상 용량의 3~20배에 해당하는 노출량으로 이브루티닙을 투여한 결과, 구조적 이상을 포함한 배자-태아 독성이 나타났습니다 (자료 참조). 임신 여성에게 태아에 대한 잠재적 위험을 알려주십시오.
모든 임신에는 기형, 유산 또는 기타 부작용의 배경 위험이 있습니다. 해당 인구집단에 대한 주요 선천적 기형 및 유산의 추정 배경 위험은 알 수 없습니다. 미국 일반 인구에서 임상적으로 인지된 임신에서 주요 선천적 기형 및 유산의 추정 배경 위험은 각각 2~4% 및 15~20%입니다.
자료
동물 자료
이브루티닙은 기관 형성 기간 동안 임신 랫트에 10, 40 및 80mg/kg/일의 용량으로 경구 투여되었습니다. 80mg/kg/일 용량의 이브루티닙은 내장 기형(심장 및 주요 혈관) 및 흡수 증가 및 착상 후 손실과 관련이 있었습니다. 랫트에서 80mg/kg/일 용량은 1일 420mg을 투여받은 CLL/SLL 또는 WM 환자의 노출량의 약 20배입니다. 40mg/kg/일 이상의 이브루티닙 용량은 태아 체중 감소와 관련이 있었습니다. 랫트에서 40mg/kg/일 용량은 1일 420mg을 투여받은 환자의 노출량(AUC)의 약 8배입니다.
이브루티닙은 기관 형성 기간 동안 임신 토끼에 5, 15 및 45mg/kg/일의 용량으로 경구 투여되었습니다. 15mg/kg/일 이상의 이브루티닙 용량은 골격 변이(융합된 흉골)와 관련이 있었고, 45mg/kg/일 용량의 이브루티닙은 흡수 증가 및 착상 후 손실과 관련이 있었습니다. 토끼에서 15mg/kg/일 용량은 1일 420mg을 투여받은 CLL/SLL 또는 WM 환자의 노출량의 약 2.8배입니다.
8.2
수유
위험 요약
모유에서 이브루티닙 또는 그 대사물질의 존재 여부, 수유아에 대한 영향 또는 모유 생산에 대한 영향에 대한 정보는 없습니다. 수유아에게 심각한 부작용이 발생할 가능성이 있으므로 IMBRUVICA 치료 중 및 마지막 투여 후 1주일 동안 여성이 수유하지 않도록 조언하십시오.
8.3
생식 잠재력이 있는 여성 및 남성
임신 여성에게 IMBRUVICA를 투여하면 태아에게 해를 끼칠 수 있습니다 [특정 인구집단에서의 사용 (8.1)] 참조.
임신 검사
IMBRUVICA 투여를 시작하기 전에 생식 잠재력이 있는 여성의 임신 상태를 확인하십시오.
피임
여성
생식 잠재력이 있는 여성에게 IMBRUVICA 치료 중 및 마지막 투여 후 1개월 동안 효과적인 피임법을 사용하도록 조언하십시오.
남성
생식 잠재력이 있는 여성 파트너가 있는 남성에게 IMBRUVICA 치료 중 및 마지막 투여 후 1개월 동안 효과적인 피임법을 사용하도록 조언하십시오.
8.4
소아 사용
만성 이식편대숙주병(cGVHD)
1세 이상의 소아 환자에서 전신 치료 1회 이상 실패 후 cGVHD 치료에 대한 IMBRUVICA의 안전성 및 유효성이 입증되었습니다.
이 적응증에 대한 IMBRUVICA의 사용은 이전에 치료받은 cGVHD가 있는 1세 이상의 소아 환자를 포함한 iMAGINE 연구(소아 환자 연령대: 1세 이상 2세 미만 1명, 2세 이상 12세 미만 20명, 12세 이상 17세 미만 19명)의 증거에 의해 뒷받침됩니다. 성인에 대한 추가적인 유효성 데이터는 연구 1129에서 제공되었습니다 [부작용 (6.1), 임상 약리학 (12.3), 및 임상 연구 (14.3)].
12세 이상 환자의 권장 IMBRUVICA 용량은 성인과 동일하며, 1세 이상 12세 미만 환자의 권장 용량은 체표면적(BSA)을 기준으로 합니다 [용법 및 용량 (2.1)].
1세 미만 소아 환자의 경우 이 적응증에 대한 IMBRUVICA의 안전성 및 유효성이 입증되지 않았습니다.
성숙 B 세포 비호지킨 림프종
이전에 치료받은 성숙 B 세포 비호지킨 림프종이 있는 35명의 환자(소아 환자 26명, 5세 이상 17세 미만)를 대상으로 한 공개, 무작위 연구(NCT02703272)를 기반으로 화학 면역 요법과 병용한 IMBRUVICA의 안전성 및 유효성이 평가되었지만 입증되지 않았습니다. 이 연구는 무효성으로 중단되었습니다. 무작위 배정된 인구집단에서 이브루티닙 + 화학 면역 요법군은 화학 면역 요법 단독군에 비해 주요 출혈 및 부작용으로 인한 화학 면역 요법 중단이 더 자주 발생했습니다.
CLL/SLL, 17p 결손이 있는 CLL/SLL, WM
소아 환자에서 CLL/SLL, 17p 결손이 있는 CLL/SLL 또는 WM에 대한 IMBRUVICA의 안전성 및 유효성은 입증되지 않았습니다.
![이브루티닙의 다음 구조는 키나제 저해제입니다. 백색에서 백색을 띠는 고체이며, 실험식은 C25H24N6O2이고 분자량은 440.50입니다. 이브루티닙은 디메틸 설폭사이드에 자유롭게 용해되고, 메탄올에 용해되며, 물에는 거의 용해되지 않습니다. 이브루티닙의 화학적 명칭은 1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one이며,](/images/44/imbruvica-01.jpg)