의약품 제조업체: Apotex Corp. (Updated: 2024-04-22)
사용상 주의사항 요약
GEFITINIB 정제, 경구 사용
최초 미국 승인: 2015
사용 적응증
용량 및 투여 방법
권장 용량은 식사 여부와 관계없이 1일 1회 250mg 경구 투여입니다. (2.2)
제형 및 강도
정제: 250mg. (3)
금기사항
없음. (4)
경고 및 주의사항
- •
- 간질성 폐렴(ILD): Gefitinib 정제 복용 환자에서 ILD가 발생했습니다. 호흡기 증상이 악화되면 Gefitinib 정제 투여를 중지하십시오. ILD가 확인되면 Gefitinib 정제 투여를 중단하십시오. (2.4, 5.1)
- •
- 간독성: 정기적으로 간기능 검사를 실시하십시오. ALT 및/또는 AST 상승이 2등급 이상인 경우 Gefitinib 정제 투여를 중지하십시오. 중증 간장애가 있는 경우 투여를 중단하십시오. (2.4, 5.2)
- •
- 위장관 천공: 위장관 천공이 있는 경우 Gefitinib 정제 투여를 중단하십시오. (2.4, 5.3)
- •
- 설사: 3등급 이상의 설사가 있는 경우 Gefitinib 정제 투여를 중지하십시오. (2.4, 5.4)
- •
- 각막염 등 안과 질환: 중증 또는 악화된 안과 질환 징후 및 증상이 있는 경우 Gefitinib 정제 투여를 중지하십시오. 지속적인 궤양성 각막염이 있는 경우 Gefitinib 정제 투여를 중단하십시오. (2.4, 5.5)
- •
- 수포성 및 박탈성 피부질환: 3등급 이상의 피부 반응 또는 박탈성 피부질환이 있는 경우 Gefitinib 정제 투여를 중지하십시오. (2.4, 5.6)
- •
- 배아/태아 독성: 태아에게 해를 줄 수 있습니다. 태아에 대한 잠재적 위험과 효과적인 피임법 사용에 대해 상담하십시오. (5.7, 8.1, 8.3)
이상반응
환자의 20% 이상에서 발생하고 위약군보다 높은 비율로 보고된 가장 흔한 이상반응은 피부 반응과 설사였습니다. (6.1)
의심되는 이상반응은 Apotex Inc.(1-800-706-5575)나 FDA(1-800-FDA-1088 또는 www.fda.gov/medwatch)에 보고하십시오.
약물 상호작용
특정 인구집단에서의 사용
- •
- 모유 수유: 수유를 중단하십시오. (8.2)
환자 상담 정보 및 FDA 승인 환자 설명서는 17항을 참조하십시오.
개정: 2023년 8월
목차
전체 처방 정보: 목차*
1 적응증 및 용법
2 투여량 및 투여방법
2.1 환자 선별
2.2 권장 투여량
2.3 고형물 삼키기 어려운 환자에 대한 투여
2.4 투여량 조절
3 제형 및 강도
4 금기사항
5 경고 및 주의사항
5.1 간질성 폐질환(ILD)
5.2 간독성
5.3 위장관 천공
5.4 중증 또는 지속적인 설사
5.5 각막염 등의 안구 장애
5.6 수포성 및 박리성 피부 장애
5.7 태아 독성
6 이상반응
6.1 임상시험 경험
6.2 시판 후 경험
7 약물 상호작용
7.1 게피티닙 노출에 영향을 미치는 약물
7.2 와파린 복용 환자에서의 출혈
8 특정 집단에서의 사용
8.1 임신
8.2 모유 수유
8.3 생식 가능 여성 및 남성
8.4 소아 환자
8.5 고령 환자
8.6 신장애 환자
8.7 간장애 환자
10 과량투여
11 설명
12 임상 약리학
12.1 작용 기전
12.3 약동학
13 비임상 독성 시험
13.1 발암성, 돌연변이성, 생식능력 손상
14 임상 연구
16 공급 형태/보관 및 취급 방법
17 환자 상담 정보
- *
- 전체 처방 정보에서 생략된 절 또는 절차는 포함되지 않습니다.
1 적응증 및 용법
게피티닙 정제는 FDA 승인 검사에서 검출된 상피 성장 인자 수용체(EGFR) 엑손 19 결실 또는 엑손 21(L858R) 치환 돌연변이를 가진 전이성 비소세포폐암 환자의 초회 치료 적응증이 있습니다 [임상 연구(14) 참조]
사용 제한: 게피티닙 정제의 안전성과 유효성은 엑손 19 결실 또는 엑손 21(L858R) 치환 돌연변이 외의 EGFR 돌연변이를 가진 전이성 비소세포폐암 환자에서 확립되지 않았습니다 [임상 연구(14) 참조]
2 용량 및 투여법
2.1 환자 선별
전이성 비소세포폐암의 1차 치료를 위해 종양 또는 혈장 검체에서 EGFR 엑손 19 결손 또는 엑손 21 L858R 돌연변이가 있는 환자를 게피티닙 정제로 선별하십시오.[사용 적응증(1), 임상 연구(14)] 혈장 검체에서 이러한 돌연변이가 검출되지 않으면 가능한 경우 종양 조직을 검사하십시오.
비소세포폐암에서 EGFR 돌연변이 검출을 위한 FDA 승인 검사에 대한 정보는 http://www.fda.gov/CompanionDiagnostics에서 확인할 수 있습니다.
2.2 권장 용량
게피티닙 정제의 권장 용량은 식사와 관계없이 매일 1회 250mg을 경구 투여하며, 질병 진행 또는 허용할 수 없는 독성이 나타날 때까지 투여합니다.
다음 복용 시간 12시간 이내에는 놓친 복용량을 섭취하지 마십시오.
2.3 고형물 삼키기 어려운 환자에 대한 투여법
4~8온스의 물에 게피티닙 정제를 담그고 약 15분간 저어줍니다. 그 후 바로 마시거나 비위관을 통해 투여합니다. 마실 때 4~8온스의 물로 용기를 헹구고 바로 마시거나 비위관을 통해 투여합니다.
2.4 용량 조절
이상반응에 대한 용량 조절
다음과 같은 경우 게피티닙 정제 투여를 중지합니다(최대 14일):
- •
- 폐 증상(호흡곤란, 기침, 발열)의 급성 발현 또는 악화[경고 및 주의사항(5.1)참조]
- •
- NCI CTCAE 등급 2 이상의 ALT 및/또는 AST 상승[경고 및 주의사항(5.2)참조]
- •
- NCI CTCAE 등급 3 이상의 설사[경고 및 주의사항(5.4)참조]
- •
- 각막염을 포함한 중증 또는 악화된 안구 장애의 징후 및 증상[경고 및 주의사항(5.5)참조]
- •
- NCI CTCAE 등급 3 이상의 피부 반응[경고 및 주의사항(5.6)참조]
이상반응이 완전히 해결되거나 NCI CTCAE 등급 1로 개선되면 게피티닙 정제 투여를 재개하십시오.
다음과 같은 경우 게피티닙 정제 투여를 영구적으로 중단하십시오:
- •
- 간질성폐렴(ILD) 확인[경고 및 주의사항(5.1)참조]
- •
- 중증 간장애[경고 및 주의사항(5.2)참조]
- •
- 위장관 천공[경고 및 주의사항(5.3)참조]
- •
- 지속되는 궤양성 각막염[경고 및 주의사항(5.5)참조]
약물상호작용에 대한 용량 조절
강력한 CYP3A4 유도제
중대한 이상반응이 없다면 게피티닙 정제 용량을 500mg으로 증량하고, 강력한 CYP3A4 유도제 중단 7일 후 250mg으로 돌아갑니다[약물상호작용(7)참조, 임상 약리학(12.3)참조].
3 투여 형태 및 강도
250mg 정제: 붉은 갈색의 원형 양면체 코팅 정제입니다. 한 면에는 “APO”가 새겨져 있고 다른 면에는 “250”이 새겨져 있습니다.
4 금기 사항
없음.
5 경고 및 주의사항
5.1 간질성 폐질환(ILD)
임상 시험에서 2462명의 게피티닙 투여 환자 중 1.3%에서 ILD 또는 ILD 유사 이상 약물 반응(예: 폐 침윤, 폐렴, 급성 호흡곤란 증후군 또는 폐 섬유증)이 발생했으며 이 중 0.7%는 3등급 이상이었고 3건이 치명적이었습니다.
호흡 곤란, 기침 및 발열과 같은 호흡기 증상이 악화된 환자에서는 게피티닙 정제를 중단하고 즉시 ILD 여부를 조사해야 합니다. ILD가 확인되면 게피티닙 정제를 영구적으로 중단해야 합니다[투여량 및 투여방법(2.4), 이상반응(6.1)를 참조하십시오].
5.2 간독성
임상 시험에서 게피티닙을 투여받은 환자 중 11.4%에서 알라닌 아미노전이효소(ALT) 수치가 상승했고, 7.9%에서 아스파르테이트 아미노전이효소(AST) 수치가 상승했으며, 2.7%에서 빌리루빈 수치가 상승했습니다. ALT 5.1%, AST 3.0%, 빌리루빈 0.7%의 환자에서 3등급 이상의 간기능 이상이 발생했습니다. 치명적 간독성의 발생률은 0.04%였습니다.
주기적인 간기능 검사를 시행해야 합니다. 간기능 악화 환자에서는 게피티닙 정제를 중단하고 중증 간장애 환자에서는 중단해야 합니다 [투여량 및 투여방법(2.4), 이상반응(6.1), 특정 집단에서의 사용(8.7)참조].
5.3 위장관 천공
임상 시험에서 2462명의 게피티닙 투여 환자 중 3명(0.1%)에서 위장관 천공이 발생했습니다[이상반응(6.1) 참조]. 위장관 천공이 발생하면 게피티닙 정제를 영구적으로 중단해야 합니다[투여량 및 투여방법(2.4) 참조].
5.4 중증 또는 지속적 설사
임상 시험에서 2462명의 게피티닙 투여 환자 중 3%에서 3 또는 4등급 설사가 발생했습니다. 중증 또는 지속적(최대 14일까지) 설사가 있는 경우 게피티닙 정제를 중단해야 합니다 [투여량 및 투여방법(2.4) 및 이상반응(6.1)참조].
5.5 각막염 포함 안과 질환
임상 시험에서 2462명의 게피티닙 투여 환자 중 각막염(0.1%), 각막 궤양 및 비정상적 속눈썹 성장(0.2%), 결막염, 안검염 및 건조증(6.7%)과 같은 안과 질환이 발생했습니다. 3등급 안과 질환의 발생률은 0.1%였습니다[이상반응(6.1) 참조]. 중증 또는 악화되는 안과 질환이 있는 경우 게피티닙 정제 투여를 중단하거나 중단해야 합니다[투여량 및 투여방법(2.4) 참조].
5.6 수포성 및 박탈성 피부 질환
독성 표피 괴사증, 스티븐스-존슨 증후군 및 다형 홍반과 같은 수포성 질환이 게피티닙 치료 중 보고되었습니다. 비소세포폐암 시험(연구 2, 연구 3 및 연구 4)에서 2명(0.08%)의 환자에서 다형 홍반 및 수포성 피부염이 보고되었습니다. 중증 수포, 수포성 또는 박탈성 질환이 있는 경우 게피티닙 정제 치료를 중단하거나 중단해야 합니다.
5.7 태아 독성
작용 기전 및 동물 생식 연구 데이터에 따르면 게피티닙 정제는 임신한 여성에게 투여할 경우 태아에 해를 줄 수 있습니다. 동물 생식 연구에서 경구 투여 시 권장 인체 투여량 미만의 용량에서 태아독성 및 신생아 사망이 발생했습니다. 임신부에게 태아 위험 가능성을 알려야 합니다. 가임 여성에게는 게피티닙 정제 투여 중 및 투여 완료 후 최소 2주 동안 효과적인 피임법을 사용할 것을 권고합니다[특정 집단에서의 사용(8.1, 8.3)참조].
부작용
다음과 같은 부작용은 라벨의 다른 섹션에서 더 자세히 논의됩니다:
- •
- 간질성 폐 질환 [경고 및 주의사항 (5.1) 참조]
- •
- 간독성 [경고 및 주의사항 (5.2) 참조]
- •
- 위장관 천공 [경고 및 주의사항 (5.3) 참조]
- •
- 심각하거나 지속적인 설사 [경고 및 주의사항 (5.4) 참조]
- •
- 각막염을 포함한 안구 질환 [경고 및 주의사항 (5.5) 참조]
- •
- 수포성 및 박리성 피부 질환 [경고 및 주의사항 (5.6) 참조]
6.1 임상시험 경험
임상시험은 다양한 조건에서 실시되므로, 한 약물의 임상시험에서 관찰된 부작용 발생률을 다른 약물의 임상시험 결과와 직접 비교할 수 없으며, 실제 관찰률과 다를 수 있습니다.
게피티닙 정제의 안전성은 3개의 무작위 임상시험(2차 연구, 3차 연구 및 4차 연구)에서 게피티닙 250mg 단일요법을 투여받은 2,462명의 비소세포폐암 환자로부터 얻은 데이터에 기반합니다. 이전에 간질성 폐 질환, 약물유발성 간질성 질환, 스테로이드 치료가 필요한 방사선 폐렴 또는 임상적으로 활성화된 간질성 폐 질환이 있었던 환자는 이러한 연구에서 제외되었습니다.
대조군 연구:
2차 연구는 다기관 공개 임상시험으로, 1,217명의 환자가 전이성 비소세포폐암에 대한 1차 치료제로 무작위 배정되었습니다. 607명의 환자가 게피티닙 250mg을 매일 투여받았고, 589명의 환자가 카보플라틴/파클리탁셀을 투여받았습니다. 게피티닙 치료 기간의 중앙값은 5.9개월이었습니다. 연구 대상 환자군의 특성은 다음과 같습니다: 연령 중앙값 57세, 65세 미만(73%), 여성(79%), 아시아인(100%), 비소세포폐암 선암종 조직형(100%), 비흡연자(94%), 경흡연자(6%), 0 또는 1의 ECOG 수행상태 등급(90%).
3차 연구는 다기관 이중맹검 위약대조 임상시험으로, 1,692명의 환자가 전이성 비소세포폐암에 대한 2차 또는 3차 치료제로 무작위 배정되었습니다. 그중 1,126명의 환자가 게피티닙 250mg을 매일 투여받았고, 562명의 환자가 위약을 투여받았습니다. 게피티닙 치료 기간의 중앙값은 2.9개월이었습니다. 연구 대상 환자군의 특성은 다음과 같습니다: 연령 중앙값 62세, 65세 미만(60%), 여성(33%), 백인(75%), 아시아인(21%), 비소세포폐암 선암종 조직형(48%), 비흡연자(22%), 0 또는 1의 ECOG 수행상태 등급(65%), 2의 수행상태 등급(29%), 3의 수행상태 등급(5%), 그리고 두 가지 이상의 이전 치료(51%).
4차 연구는 다기관 공개 임상시험으로, 1,466명의 환자가 전이성 비소세포폐암에 대한 2차 치료제로 무작위 배정되었습니다. 729명의 환자가 게피티닙 250mg을 매일 투여받았고, 715명의 환자가 독세탁셀을 투여받았습니다. 게피티닙 정제 치료 기간의 중앙값은 2.4개월이었습니다. 연구 대상 환자군의 특성은 다음과 같습니다: 연령 중앙값 61세, 65세 미만(61%), 여성(36%), 백인(79%), 아시아인(21%), 비소세포폐암 선암종 조직형(54%), 비흡연자(20%), 0 또는 1의 ECOG 수행상태 등급(88%), 그리고 두 가지 이상의 이전 치료(16%).
심각하고 드문 부작용을 평가하기 위해 세 가지 무작위 임상시험의 통합 안전성 데이터베이스를 사용했습니다. 흔한 부작용은 3차 연구에서 평가되었습니다. 게피티닙 투여 환자에서 가장 빈번한 부작용(발생률 20% 이상이며 위약군보다 높음)은 피부 반응(47%) 및 설사(29%)였습니다. 게피티닙 투여 환자에서 가장 빈번한 치명적인 부작용은 호흡기 부전(0.9%), 폐렴(0.8%) 및 폐색전증(0.5%)이었습니다.
게피티닙 투여군의 약 5%, 위약군의 2.3%가 부작용으로 인해 치료를 중단했습니다. 게피티닙 투여 환자에서 치료 중단을 초래한 가장 흔한 부작용은 구역질(0.5%), 구토(0.5%) 및 설사(0.4%)였습니다.
표 1 – 3차 연구에서 발생률 ≥5% 이면서 게피티닙 투여군에서 2% 이상 증가한 선별된 부작용
| 부작용 | 발생 환자 비율(%) | |||
| 게피티닙 (N=1126) | 위약 (N=562) | |||
| 전체 등급 | 3&4등급 | 전체 등급 | 3&4등급 | |
| 피부 및 피하 조직 장애 | ||||
| 피부 반응1 | 47% | 2% | 17% | 0.4% |
| 손발톱 장애2 | 5% | 0.1% | 0.7% | 0% |
| 위장관 장애 | ||||
| 설사3 | 29% | 3% | 10% | 1% |
| 구토 | 14% | 1.2% | 10% | 0.4% |
| 구내염4 | 7% | 0.3% | 4% | 0.2% |
| 대사 및 영양 장애 | ||||
| 식욕감퇴 | 17% | 2.3% | 14% | 2% |
| 안구 장애 | ||||
| 결막염/안검염/건조 증상5 | 6% | 0% | 3.2% | 0% |
1 여드름, 농포성 여드름, 피부염, 주사 피부염, 박리성 피부염, 약물 발진, 건조 피부, 홍반, 박리성 발진, 모낭염, 가려움, 전신 가려움, 발진, 홍반성 발진, 전신 발진, 반구진 발진, 구진 발진, 소양증 발진, 농포성 발진, 수포성 발진, 피부 박리, 피부 독성, 건피증 포함
2 내향성 발톱, 발톱 주변 염증, 발톱 장애, 발톱 감염, 발톱 파열, 발톱 박리, 지휘염 포함
3 설사, 연변, 잦은 대변 포함
4 구내염, 구순염, 설통, 구내염, 점막염, 구강 점막 수포, 구내염, 설질환, 설궤양 포함
5 결막염, 결막 충혈, 결막염, 건성안, 안자극, 눈가려움증, 안구 부종, 눈꺼풀 자극, 눈꺼풀 부종, 눈꺼풀 가려움증 포함
표 2 – 3차 연구에서 Gefitinib 치료군에서 더 자주 발생한 실험실 검사 이상
| 이상반응 | Gefitinib | 위약 | ||
| 전체 등급% | 3 및 4등급% | 전체 등급% | 3 및 4등급% | |
| 알라닌 아미노전이효소 증가1 | 38%2 | 2.4% | 23%2 | 1.4%4 |
| 아스파르테이트 아미노전이효소 증가1 | 40%3 | 2% | 25%3 | 1.3%5 |
| 단백뇨 | 35% | 4.7% | 31% | 3.3% |
1 환자는 ALT 또는 AST CTCAE 1 또는 2등급 수치로 임상시험에 참여할 수 있었음
2 Gefitinib군 14%, 위약군 10%가 기저치 ALT 1 또는 2등급
3 Gefitinib군 15%, 위약군 12%가 기저치 AST 1 또는 2등급
4 위약군 0.2%가 기저치 3등급
5 위약군 0.4%가 기저치 3등급
다음 이상반응은 비소세포폐암 임상시험(2차, 3차, 4차 연구)에서 gefitinib과 관련하여 보고되었지만 6절의 다른 곳에는 나열되지 않았다: 구역(18%), 권태(17%), 발열(9%), 탈모(4.7%), 출혈(비출혈 및 혈뇨 포함)(4.3%), 구강건조(2%), 탈수(1.8%), 혈중 크레아티닌 상승(1.5%), 혈관부종 및 두드러기를 포함한 알레르기 반응(1.1%), 손발농피증(0.2%) 및 췌장염(0.1%).
6.2 시판 후 경험
다음 이상반응은 gefitinib의 허가 후 사용 중에 확인되었다. 이러한 반응이 불확실한 규모의 인구에서 자발적으로 보고되었기 때문에 항상 그 빈도를 신뢰성 있게 추정하거나 약물 노출과의 인과관계를 확립하는 것은 불가능하다.
신장 및 비뇨기계 장애: 방광염, 출혈성 방광염
피부 및 피하조직 장애: 피부 혈관염
7 약물 상호작용
7.1 게피티닙 노출에 영향을 미치는 약물
CYP3A4 유도제
CYP3A4의 강력한 유도제는 게피티닙의 대사를 증가시키고 게피티닙의 혈장 농도를 낮춥니다. CYP3A4의 강력한 유도제(예: 리팜핀, 페니토인 또는 삼환계 항우울제)를 투여받는 환자의 경우 게피티닙 정제를 하루 500mg으로 증량하고, 강력한 유도제 투여 중단 7일 후에는 게피티닙 정제를 250mg으로 복귀시킵니다.[용법 및 투여량 (2.4), 임상약리학 (12.3)].
CYP3A4 억제제
CYP3A4의 강력한 억제제(예: 케토코나졸 및 이트라코나졸)는 게피티닙의 대사를 감소시키고 게피티닙의 혈장 농도를 높입니다. CYP3A4 강력한 억제제를 게피티닙 정제와 함께 투여할 때 부작용을 모니터링하십시오.
위 산도에 영향을 미치는 약물
위 산도를 높이는 약물(예: 프로톤 펌프 억제제, 히스타민 H2-수용체 길항제 및 제산제)은 게피티닙의 혈장 농도를 낮출 수 있습니다. 가능하면 게피티닙 정제와 프로톤 펌프 억제제를 병용하지 마십시오. 프로톤 펌프 억제제 치료가 필요한 경우 프로톤 펌프 억제제의 마지막 투여 12시간 후 또는 다음 투여 12시간 전에 게피티닙 정제를 복용하십시오. H2-수용체 길항제 또는 제산제 투여 6시간 후 또는 6시간 전에 게피티닙 정제를 복용하십시오[임상약리학 (12.3)].
7.2 와파린 복용 환자에서의 출혈
와파린을 투여받는 일부 환자에서 게피티닙 요법 중 국제 표준화 비율(INR) 상승 및/또는 출혈이 보고되었습니다. 와파린을 복용하는 환자의 경우 프로트롬빈 시간 또는 INR 변화를 정기적으로 모니터링해야 합니다.
8 특정 인구에서의 사용
8.1 임신
위험 요약
작용 기전과 동물 데이터를 기반으로 할 때, 제피티닙 정제는 임신 중 투여 시 태아에게 해로울 수 있습니다. 동물 생식 연구에서, 제피티닙의 경구 투여(기관 발생기부터 젖떼는 때까지)는 권장 인체 투여량보다 낮은 용량에서 태아독성 및 신생아 사망을 초래했습니다(동물 데이터를 참조하십시오). 임신부에게 태아 위험 또는 임신 손실 위험이 있을 수 있음을 알립니다.
대상 인구의 주요 선천성 기형 및 유산 위험율은 알려져 있지 않지만, 미국 일반 인구의 주요 선천성 기형 위험은 2~4%, 유산 위험은 임상적으로 확인된 임신의 15~20%입니다.
데이터
동물 데이터
랫트 단회 투여 연구에서 제피티닙은 5 mg/kg(30 mg/m2, 인체 권장 투여량의 약 0.2배) 경구 투여 후 태반을 통과함을 보였습니다. 기관 발생기부터 젖떼는 때까지 임신 랫트에게 5 mg/kg를 투여했을 때 생존한 새끼 수가 감소했습니다. 이 효과는 20 mg/kg(인체 임상 투여량과 대략 동일한 mg/m2 기준)에서 더 심각했고, 분만 직후 신생아 사망률이 높았습니다. 토끼에서 20 mg/kg/일(240 mg/m2, 인체 권장 투여량의 약 2배)의 용량은 태아 체중 감소를 초래했습니다.
8.2 모유 수유
위험 요약
제피티닙이 인체 모유로 배설되는지는 알려져 있지 않습니다. 동물 연구에서는 제피티닙과 그 대사체가 모체 혈장보다 높은 농도로 랫트 젖에 존재함을 보였습니다. 제피티닙으로 인한 수유아의 심각한 이상반응 위험 때문에, 제피티닙 정제 치료 기간 동안 모유 수유를 중단할 것을 권합니다.
데이터
동물 데이터
수유 랫트에게 5 mg/kg 경구 투여 후, 제피티닙과 대사체의 농도는 젖에서 혈액보다 11~19배 높았습니다.
8.3 생식 연령 여성 및 남성
피임 요법
작용 기전과 동물 데이터에 근거할 때, 제피티닙 정제는 임신부에게 투여되면 태아에게 해로울 수 있습니다[특정 인구에서의 사용(8.1)을 참조하십시오]. 생식 연령 여성에게 제피티닙 정제 치료 기간과 치료 종료 후 2주 동안 효과적인 피임을 할 것을 권합니다.
불임
제피티닙 정제는 생식 연령 여성의 불임을 초래할 수 있습니다[비임상 독성 연구(13.1)를 참조하십시오].
8.5 고령 환자 사용
무작위 배정, 활성 대조 임상시험 두 건에 등록된 823명 환자 중 374명(45%)이 65세 이상이었고, 93명(11%)이 75세 이상이었습니다. 65세 이상 환자와 65세 미만 환자 간에 전반적인 안전성 차이는 관찰되지 않았습니다. 고령 환자와 젊은 환자 사이의 유효성 차이를 평가하기에는 정보가 충분하지 않습니다.
8.7 간장애
간경화(Child-Pugh 분류에 따름)로 인한 경증, 중등증 또는 중증 간장애 환자와 정상 간기능 건강 대상군(각 군 10명)에서 제피티닙의 전신 노출량을 비교했습니다. 평균 전신 노출량(AUC0-∞)은 경증 장애 환자에서 40%, 중등증 장애 환자에서 263%, 중증 간장애 환자에서 166% 증가했습니다. 제피티닙 정제를 중등증 및 중증 간장애 환자에게 투여할 때에는 이상반응을 모니터링합니다.
중등도의 간 기능 장애(기저 AST/SGOT, ALP 및 빌리루빈 CTC 등급의 합이 3~5인 경우)를 가진 간전이 환자 13명과 정상 간기능 간전이 환자 14명을 비교한 연구에서, 제피티닙의 전신 노출량은 유사했습니다[경고 및 주의사항(5.2)를 참조하십시오].
10 과다 복용
23명의 환자가 1500mg에서 3500mg 사이의 용량으로 매주 치료를 받았으며, 게피티닙 노출 정도가 용량 증가와 함께 증가하지 않았습니다. 부작용은 대부분 경증에서 중등도였으며 게피티닙의 알려진 안전성 프로파일과 일치했습니다. 과다 복용이 의심되는 경우 게피티닙 정제 복용을 중단하고, 보조 치료를 실시하며 임상적으로 안정될 때까지 관찰합니다. 게피티닙 정제의 과다 복용 후 취해야 할 특정 조치/치료는 없습니다.
11 설명
게피티닙은 키나제 억제제입니다.
게피티닙의 화학명은 4-퀴나졸린아민 N-(3-클로로-4-플루오로페닐)-7-메톡시-6-[3-(4-모르폴리닐)프로폭시]이고 다음과 같은 구조식을 가집니다:

게피티닙의 분자식은 C22H24ClFN4O3이며, 상대 분자량은 446.91 달톤이고 흰색 또는 회백색 가루입니다. 게피티닙은 유리염기입니다. 이 분자는 pKa가 5.4와 7.2입니다. 게피티닙은 pH 1에서 약간 용해되지만, pH 7 이상에서는 실제로 불용성이며, pH 4와 pH 6 사이에서 용해도가 급격히 감소합니다. 비수용성 용매에서 게피티닙은 빙초산과 디메틸술폭시드에 자유롭게 용해되고, 피리딘에 용해되며, 테트라히드로푸란에 약간 용해되고, 메탄올, 에탄올(99.5%), 에틸아세테이트, 프로판-2-올 및 아세토니트릴에 약간 용해됩니다.
게피티닙 정제는 경구 투여용 250 mg의 게피티닙을 함유하는 갈색, 원형, 양면볼록 코팅 정제입니다. 게피티닙 정제의 비활성 성분은 아미노메타크릴레이트 공중합체, 콜로이드성 이산화규소, 크로스포비돈 및 스테아린산 마그네슘입니다. 정제 코팅은 적색 산화철, 폴리에틸렌글리콜, 폴리비닐알코올, 탈크 및 이산화티탄으로 구성되어 있습니다.
12 임상 약리학
12.1 작용 기전
상피 성장 인자 수용체(EGFR)는 정상 세포와 암 세포 표면에 존재하며 세포 성장 및 증식 과정에 관여합니다. 일부 EGFR 활성화 돌연변이(엑손 19 결실 또는 엑손 21 점 돌연변이 L858R)는 비소세포폐암 세포에서 발견되었으며, 이는 종양 세포 성장 촉진, 세포자멸사 차단, 혈관 생성 인자 생산 증가 및 전이 과정 촉진에 기여하는 것으로 알려져 있습니다.
게피티닙은 야생형 및 특정 활성화 EGFR 돌연변이의 키나제 활성을 가역적으로 억제하여 수용체와 관련된 티로신 잔기의 자가인산화를 방지하고 추가적인 하위 신호 전달을 차단하여 EGFR 의존적인 증식을 억제합니다.
게피티닙은 EGFR 엑손 19 결실 또는 엑손 21 점 돌연변이 L858R에 대한 결합 친화력이 야생형 EGFR에 대한 결합 친화력보다 높습니다. 또한 게피티닙은 임상적으로 관련 있는 농도에서 IGF 및 PDGF 매개 신호 전달을 억제합니다. 다른 티로신 키나제 수용체에 대한 억제 작용은 완전히 확인되지 않았습니다.
12.3 약동학
흡수 및 분포
게피티닙의 평균 경구 생체이용률은 60%이며 최고 혈장 농도에 도달하는 시간은 투여 후 3~7시간입니다. 음식물은 게피티닙의 생체이용률에 임상적으로 유의한 영향을 주지 않습니다. 게피티닙 정제는 음식물과 함께 또는 따로 복용할 수 있습니다. 게피티닙은 정맥 투여 후 평균 정상 상태 분포 용적이 1400L로 신체 전체에 광범위하게 분포합니다. In vitro에서 게피티닙의 인체 혈장 단백질(혈청 알부민 및 α1-산 글리코단백질) 결합률은 90%이며 약물 농도에 관계없습니다. 게피티닙은 막 수송체 P-glycoprotein(P-gp)의 기질이지만 높은 농도에서 P-gp가 포화되기 때문에 흡수에 영향을 미치지 않을 것으로 보입니다.
대사 및 제거
게피티닙은 사람에서 주로 CYP3A4에 의해 광범위한 간 대사를 거칩니다. 세 가지 생물전환 부위가 확인되었습니다: N-프로폭시모르폴리노기의 대사, 퀴나졸린의 메톡시기 탈메틸화, 할로겐화 페닐기의 산화적 탈불소화입니다. 분변 추출물에서 다섯 가지 대사체가 완전히 동정되었으며, CYP2D6 대사에 의해 생성된 O-데스메틸 게피티닙이 주요 활성 성분이었고 용량의 14%를 차지했습니다.
인체 혈장에서 8가지 대사체가 확인되었습니다. O-데스메틸 게피티닙만이 게피티닙과 유사한 노출 수준을 보였습니다. 이 대사체는 분리된 효소 분석에서 게피티닙과 유사한 EGFR-TK 활성을 갖지만, 세포 기반 분석 중 하나에서 게피티닙의 활성이 1/14 수준이었습니다.
게피티닙은 주로 간에서 제거되며, 정맥 투여 후 총 혈장 클리어런스와 배설 반감기는 48시간입니다. 건강한 대상자에서 AUC의 개체 간 변동 계수는 67%였습니다. 암 환자에게 게피티닙을 매일 경구 투여하면 단회 투여에 비해 2배의 축적이 나타났습니다. 정상 상태 혈장 농도는 매일 투여 후 10일 이내에 도달합니다. 게피티닙과 그 대사체의 배설은 주로 분변(86%)을 통해 이루어지며, 신장을 통한 배설은 투여량의 4% 미만입니다.
특정 집단
나이, 성별, 체중, 인종 또는 신장 기능: 인구 약동학 분석 결과에 따르면 환자의 나이, 체중, 인종(연구 대상 인구 포함) 또는 크레아티닌 클리어런스(20 mL/min 초과)는 예측된 게피티닙의 정상 상태 트라프 농도에 임상적으로 유의한 영향을 주지 않습니다. 1차 연구의 인구 약동학 분석에서 여성의 노출이 남성보다 27% 높았지만, 다른 게피티닙 임상시험의 분석에서는 이러한 차이가 확인되지 않았습니다. 환자의 성별에 따른 용량 조절은 권장되지 않습니다.
간 장애: 간경화로 인한 경증, 중등증 또는 중증 간 장애 환자와 정상 간 기능 건강한 대상자(각 그룹 N=10)의 게피티닙 전신 노출을 비교했습니다. 평균 전신 노출(AUC0-∞)은 경증 장애 환자에서 40%, 중등증 장애 환자에서 263%, 중증 간 장애 환자에서 166% 증가했습니다. 간 전이가 있는 중등증 간 장애 환자 13명과 정상 간 기능 환자 14명을 비교한 연구에서 게피티닙의 전신 노출은 유사했습니다[[경고 및 주의사항(5.2), 특정 집단에서의 사용(8.7) 참조]].
CYP2D6 저대사자: CYP2D6는 in vitro에서 게피티닙을 O-데스메틸 게피티닙으로 대사시킵니다. 건강한 CYP2D6 저대사자에서는 O-데스메틸 게피티닙의 농도를 측정할 수 없었고 광범위 대사자에 비해 게피티닙 노출이 2배 높았습니다. 이러한 CYP2D6 저대사자의 노출 증가는 일부 이상반응이 게피티닙 고노출과 관련되어 있기 때문에 임상적으로 중요할 수 있습니다. CYP2D6 저대사 유전형이 알려진 환자에서는 용량 조절이 권장되지 않지만 이상반응에 대해 주의 깊게 모니터링해야 합니다. CYP2D6 억제제가 게피티닙 약동학에 미치는 영향은 평가되지 않았습니다. 그러나 이러한 환자에서 노출 증가 가능성이 있으므로 게피티닙과 CYP2D6 억제제를 병용 투여할 때 유사한 주의사항을 적용해야 합니다.
탐색적 노출-반응 분석 결과 게피티닙 노출이 2배 이상 증가하면 간질성 폐렴(ILD) 발생 위험이 높아지는 것으로 나타났습니다[[경고 및 주의사항(5.1) 참조]].
약물-약물 상호작용
강력한 CYP3A4 유도제:
강력한 CYP3A4 유도제인 리팜핀(600mg QD, 16일간)과 게피티닙(리팜핀 투여 10일차에 500mg 단회 투여)을 병용하면 게피티닙의 평균 AUC가 83% 감소했습니다. [투여량 및 투여방법(2.4), 약물상호작용(7) 참조]
CYP3A4 억제제:
CYP3A4 억제제인 이트라코나졸(200mg QD, 12일간)과 게피티닙(이트라코나졸 투여 4일차에 250mg 단회 투여)을 건강한 남성에게 병용 투여하면 게피티닙의 평균 AUC가 80% 증가합니다. [약물상호작용(7) 참조]
위 pH에 영향을 주는 약물:
건강한 대상자에게 고용량 라니티딘과 중탄산나트륨(위 pH를 5.0 이상으로 유지)을 병용 투여하면 게피티닙의 평균 AUC가 47% 감소합니다. [약물상호작용(7) 참조]
인간 간 미세소체 연구에서 게피티닙은 2~5000 ng/mL 농도 범위에서 CYP1A2, CYP2C9 및 CYP3A4 활성을 억제하지 않았습니다. 가장 높은 농도(5000 ng/mL)에서 게피티닙은 CYP2C19를 24%, CYP2D6를 43% 억제했습니다.
CYP2D6 기질인 메토프롤롤의 노출은 고형암 환자에게 게피티닙(500mg 1일 1회, 28일간)을 15일간 투여했을 때 30% 증가했습니다.
13 비임상 독성학
13.1 발암성, 변이원성, 생식 능력 손상
Gefitinib는 일련의 in vitro (박테리아 돌연변이, 마우스 림프종, 인간 림프구) 분석 및 in vivo 랫드 소핵 검사에서 유전 독성에 대해 시험되었습니다. 이러한 분석 조건에서 gefitinib는 유전적 손상을 유발하지 않았습니다.
마우스를 대상으로 한 2년 발암성 연구에서 270 mg/m2/day(mg/m2 기준으로 권장 1일 용량의 약 2배인 250 mg; 22주부터 375 mg/m2/day로 용량 감소)의 gefitinib를 투여한 결과 암컷에서 간세포 선종이 발생했습니다. 랫드를 대상으로 한 2년 발암성 연구에서 60 mg/m2/day(mg/m2 기준으로 권장 임상 1일 용량의 약 0.4배)의 gefitinib를 투여한 결과 암컷 랫드에서 간세포 선종과 횡경막 림프절의 혈관종/혈관육종이 발생했습니다. 이러한 발견 결과의 임상적 관련성은 알려져 있지 않습니다.
랫드를 대상으로 한 전용 생식능력 연구에서 ≥120 mg/m2 (mg/m2 기준으로 gefitinib 권장 인간 용량과 거의 동일한 수준) 용량에서 불규칙한 발정 증가, 황체 감소, 자궁 착상 및 배자 수 감소가 나타났습니다.
14. 임상 연구
비소세포폐암(NSCLC)
연구 1
EGFR 엑손 19 결실 또는 L858R 치환 돌연변이를 포함한 전이성 NSCLC 환자의 1차 치료제로서 게피티닙의 유효성과 안전성은 다기관, 단일군, 공개 임상 연구(연구 1)에서 입증되었습니다. 총 106명의 전이성 EGFR 돌연변이 양성 NSCLC로 진단받은 치료 경험이 없는 환자가 질병 진행 또는 내약성이 있을 때까지 매일 250 mg의 게피티닙을 투여받았습니다. 주요 유효성 평가변수는 RECIST v1.1에 따라 Blinded Independent Central Review(BICR) 및 연구자에 의해 평가된 객관적 반응률(ORR)이었습니다. 반응 지속 기간(DOR)은 추가 평가변수였습니다. 적격 환자는 EGFR 엑손 19 결실 또는 L858R, L861Q 또는 G719X 치환 돌연변이를 가져야 했으며 T790M 또는 S768I 돌연변이 또는 엑손 20 삽입이 없어야 했습니다. 이는 임상시험 분석법으로 전향적으로 결정되었습니다. 87명의 환자 종양 검체는 후향적으로 therascreen® EGFR RGQ PCR Kit를 사용하여 검사되었습니다.
연구 대상 집단의 특성은 다음과 같습니다: 중간 연령 65세, 75세 이상(25%), 65세 미만(49%), 백인(100%), 여성(71%), 비흡연자(64%), WHO PS 0(45%), WHO PS 1(48%), WHO PS 2(7%), 및 선암종 조직학적 형태(97%). 60명의 환자는 엑손 19 결실(65%), 29명의 환자는 L858R 치환(31%)을 가지고 있었으며, 2명의 환자는 각각 L861Q 또는 G719X 치환 돌연변이를 가진 종양을 가지고 있었습니다.
치료 기간의 중간값은 8.0개월이었습니다. 연구 1의 유효성 결과는 아래와 같이 요약됩니다.
표 3 – 연구 1의 유효성 결과
| 유효성 매개변수 | BICR1 평가 (n=106)2 |
연구자 평가 (n=106) |
| 객관적 반응률3 (95% CI) |
50% (41, 59) |
70% (61, 78) |
| 완전 반응률 | 0.9% | 1.9% |
| 부분 반응률 | 49% | 68% |
| 반응 지속 기간 중간값(개월) (95% CI) |
6 (5.6, 11.1) |
8.3 (7.6, 11.3) |
1 BICR, Blinded Independent Central Review
2 기저 시점에서 BICR에 의해 표적 병변이 검출되지 않은 17명의 환자는 비반응자로 간주되었습니다.
3 RECIST v 1.1에 의해 결정됨
EGFR 엑손 19 결실과 엑손 21 L858R 치환 돌연변이 환자에서의 반응률은 유사했습니다. G719X 치환 돌연변이를 가진 환자 2명에서 각각 최소 2.8개월과 5.6개월의 반응 지속 기간으로 2건의 부분 반응이 관찰되었습니다. L861Q 치환 돌연변이를 가진 2명 중 1명도 최소 2.8개월의 반응 지속 기간으로 부분 반응을 보였습니다.
연구 2
1차 치료를 받은 전이성 선암종 조직학적 형태의 NSCLC 환자를 대상으로 한 무작위 배정, 다기관, 공개 임상시험(연구 2)의 탐색적 분석 결과는 연구 1의 결과를 뒷받침했습니다. 환자는 1:1로 무작위 배정되어 매일 250 mg 게피티닙 경구 투여군 또는 최대 6주기의 카보플라틴/파클리탁셀 투여군에 배정되었습니다. 유효성 평가변수는 BICR에 의해 평가된 무진행 생존기간(PFS) 및 객관적 반응률(ORR)을 포함했습니다.
해당 하위 집단은 연구 1에서 사용된 것과 동일한 임상시험 분석법으로 EGFR 양성으로 판정되었고 BICR에 의한 후향적 평가를 위해 방사선 촬영 결과를 이용할 수 있었던 1217명 중 186명(15%)이었습니다. 이 하위 집단은 게피티닙 투여군 88명, 카보플라틴/파클리탁셀 투여군 98명으로 구성되어 있었습니다.
이 하위 집단의 인구 통계학적 및 기저 특성은 중간 연령 59세, 75세 이상(7%), 65세 미만(70%), 아시아인(100%), 여성(83%), 비흡연자(96%), 선암종 조직학적 형태(100%), PS 0-1(94%)이었습니다.
게피티닙 투여군의 치료 기간 중간값은 9.8개월이었습니다. 위험비는 게피티닙 투여군에서 유리했습니다[HR 0.54(95% CI: 0.38, 0.79)] . BICR에 의해 평가된 결과 게피티닙 투여군의 PFS 중간값은 10.9개월, 카보플라틴/파클리탁셀 투여군은 7.4개월이었습니다. 또한 BICR 평가에 따른 객관적 반응률은 게피티닙 투여군 67%(95% CI: 56, 77), 카보플라틴/파클리탁셀 투여군 41%(95% CI: 31, 51)였습니다. 반응 지속 기간 중간값은 게피티닙 투여군 9.6개월, 카보플라틴/파클리탁셀 투여군 5.5개월이었습니다.
16 제품 공급/보관 및 취급 방법
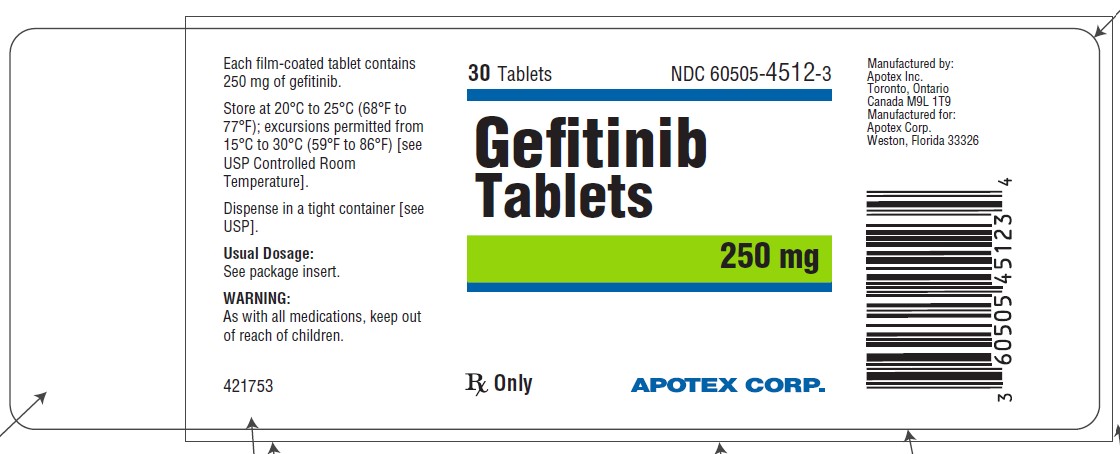
Gefitinib 정제는 250mg 정제로 제공됩니다.
Gefitinib 250mg 정제는 적갈색의 원형 양면체 코팅 정제입니다. 한쪽 면에는 “APO” 위에 “250”이 새겨져 있고 다른 한쪽 면은 평평합니다.
Gefitinib 정제는 다음과 같이 제공됩니다:
30정 병 NDC 60505-4512-3
20°C~25°C(68°F~77°F)에서 보관하며, 15°C~30°C(59°F~86°F) 범위의 온도 변화는 허용됩니다[USP 표준 실온 보관법 참조].
17 환자 상담 정보
FDA 승인 환자 라벨링(환자 정보)을 읽도록 환자에게 조언하십시오.
간질성 폐질환: 호흡곤란, 기침 및 발열과 같은 새로운 폐 증상이 발생하거나 악화되는 경우 즉시 의료 서비스 제공자에게 연락하도록 환자에게 조언하십시오[참조 경고 및 주의사항(5.1)].
간독성: 간기능을 모니터링하기 위해 검사를 받아야 한다고 환자에게 알리십시오. 새로운 간독성 증상이 나타날 경우 의료 서비스 제공자에게 연락하도록 환자에게 조언하십시오[참조 경고 및 주의사항(5.2)].
위장관 천공: 게피티닙 정제는 위장관 천공 위험을 증가시킬 수 있으므로 심한 복통이 있을 경우 즉시 의학적 주의를 구하도록 환자에게 조언하십시오[참조 경고 및 주의사항(5.3)].
심하거나 지속적인 설사: 심하거나 지속적인 설사가 있을 경우 의료 서비스 제공자에게 연락하도록 환자에게 조언하십시오[참조 경고 및 주의사항(5.4)].
각막염 등의 안구 장애: 눈물, 빛 민감성, 흐린 시력, 안구 통증, 충혈 또는 시력 변화와 같은 눈 증상이 있는 경우 즉시 의료 서비스 제공자에게 연락하도록 환자에게 조언하십시오[참조 경고 및 주의사항(5.5)]
수포성 및 박리성 피부 장애: 게피티닙 정제는 수포성 및 박리성 피부 장애의 위험을 증가시킬 수 있으므로 심각한 피부 반응이 발생할 경우 즉시 의학적 주의를 구하도록 환자에게 조언하십시오[참조 경고 및 주의사항(5.6)].
태아 독성: 가임 여성에게 태아 위험 또는 임신 손실 위험에 대해 조언하십시오[참조 경고 및 주의사항(5.7), 특정 집단에서의 사용(8.1)]. 게피티닙 정제 치료 중 및 치료 완료 후 최소 2주간 효과적인 피임법을 사용할 것을 가임 여성에게 조언하십시오[참조 특정 집단에서의 사용(8.3)].
수유: 게피티닙 정제 치료 중에는 수유를 중단할 것을 여성에게 조언하십시오[참조 특정 집단에서의 사용(8.2)].
본 문서에 있는 모든 등록 상표는 해당 소유자의 자산입니다.
APOTEX INC.
GEFITINIB TABLETS
250 mg
| 제조: | 제조 의뢰: |
| Apotex Inc. | Apotex Corp. |
| Toronto, Ontario | Weston, Florida |
| 캐나다 M9L 1T9 | 미국 33326 |
개정 7
환자 정보
주요 표시 패널
라벨링의 대표 샘플(전체 목록의 HOW SUPPLIED 섹션 참조):
APOTEX CORP.
NDC 60505-4512-3
Gefitinib Tablets
250 mg 30 카운트
Rx Only