의약품 제조업체: Kyowa Kirin, Inc. (Updated: 2024-11-01)
처방 정보의 주요 내용
CRYSVITA® (burosumab-twza) 주사, 피하 주사용
미국 최초 승인: 2018
적응증 및 사용
투여량 및 투여 방법
피하 주사 전용 (2)
-
소아 XLH(6개월 이상):
- 체중이 10kg 미만인 환자의 경우, 시작 용량 요법은 체중 1kg당 1mg을 가장 가까운 1mg으로 반올림하여 2주마다 투여합니다. (2.2)
- 체중이 10kg 이상인 환자의 경우, 시작 용량 요법은 체중 1kg당 0.8mg을 가장 가까운 10mg으로 반올림하여 2주마다 투여합니다. 최소 시작 용량은 10mg이며 최대 용량은 90mg입니다. (2.2)
혈청 인 수치를 정상화하기 위해 용량을 2주마다 약 2mg/kg(최대 90mg)까지 증가시킬 수 있습니다. (2.2)
- 성인 XLH: 용량 요법은 체중 1kg당 1mg을 가장 가까운 10mg으로 반올림하여 최대 90mg까지 4주마다 투여합니다. (2.3)
- 소아 TIO(2세 이상): 시작 용량은 체중 1kg당 0.4mg을 가장 가까운 10mg으로 반올림하여 2주마다 투여합니다. 용량은 2주마다 180mg을 초과하지 않는 범위 내에서 체중 1kg당 2mg까지 증가시킬 수 있습니다. (2.4)
- 성인 TIO: 시작 용량은 4주마다 체중 1kg당 0.5mg입니다. 용량은 2주마다 180mg을 초과하지 않는 범위 내에서 체중 1kg당 2mg까지 증가시킬 수 있습니다. (2.5)
투여 형태 및 강도
주사: 1회용 바이알에 10mg/mL, 20mg/mL 또는 30mg/mL (3)
금기 사항
경고 및 주의 사항
부작용
- 소아 XLH 환자에서 CRYSVITA군에서 가장 흔한 부작용(≥25% 및 활성 대조군 >)은 발열, 주사 부위 반응, 기침, 구토, 사지 통증, 두통, 치아 농양, 치아 우식증입니다. (6.1)
- 성인 XLH 환자에서 가장 흔한 부작용(>5% 및 위약군보다 적어도 2명 이상의 환자에서)은 요통, 두통, 치아 감염, 불안한 다리 증후군, 비타민 D 감소, 현기증, 변비, 근육 경련, 혈액 인 증가입니다. (6.1)
- TIO 환자에서 가장 흔한 부작용(>10%)은 치아 농양, 근육 경련, 현기증, 변비, 주사 부위 반응, 발진 및 두통입니다. (6.1)
의심되는 부작용을 보고하려면 Kyowa Kirin, Inc.에 1-844-768-3544 또는 FDA에 1-800-FDA-1088 또는 www.fda.gov/medwatch로 연락하십시오.
17을 참조하십시오. 환자 상담 정보.
개정: 2023년 3월
목차
FULL PRESCRIBING INFORMATION: CONTENTS*
1 적응증 및 용법
1.1 X-연관 저인산혈증
1.2 종양 유발 골연화증
2 용량 및 투여
2.1 중요 용량 및 투여 정보
2.2 X-연관 저인산혈증 소아 환자 (6개월~18세 미만)
2.3 X-연관 저인산혈증 성인 환자 (18세 이상)
2.4 종양 유발 골연화증 소아 환자 (2세~18세 미만)
2.5 종양 유발 골연화증 성인 환자 (18세 이상)
2.6 복용 누락
2.7 25-Hydroxy Vitamin D 보충
2.8 피하 투여에 대한 일반적인 고려 사항
3 제형 및 함량
4 금기
5 경고 및 주의사항
5.1 과민증
5.2 고인산혈증 및 신석회화증 위험
5.3 주사 부위 반응
6 이상반응
6.1 임상 시험 경험
6.2 면역원성
6.3 시판 후 경험
7 약물 상호작용
7.1 경구 인산염 및 활성 비타민 D 유사체
8 특정 집단에서의 사용
8.1 임신
8.2 수유
8.4 소아에서의 사용
8.5 노인에서의 사용
8.6 신장애
10 과다 복용
11 설명
12 임상 약리학
12.1 작용 기전
12.2 약력학
12.3 약동학
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 장애
13.2 동물 독성학 및/또는 약리학
14 임상 연구
14.1 소아 X-연관 저인산혈증
14.2 성인 X-연관 저인산혈증
14.3 종양 유발 골연화증
16 공급/보관 및 취급 방법
17 환자 상담 정보
- *
- 전체 처방 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1 적응증 및 용법
1.1 X-linked Hypophosphatemia
CRYSVITA는 6개월 이상의 성인 및 소아 환자의 X-linked Hypophosphatemia (XLH) 치료에 사용됩니다.
1.2 Tumor-induced Osteomalacia
CRYSVITA는 2세 이상의 성인 및 소아 환자에서 치료적으로 절제하거나 국소화할 수 없는 인산뇨성 간엽 종양과 관련된 종양 유발성 골연화증(TIO)에서 FGF23 관련 저인산혈증 치료에 사용됩니다.
2 투여 및 관리
2.1 중요한 투여 및 관리 정보
CRYSVITA 치료 시작 1주일 전에 경구 인산염 및/또는 활성 비타민 D 유사체(예: 칼시트리올, 파리칼시트리올, 닥서칼시페롤, 칼시페디올)를 중단하십시오 [금기 사항 (4) 참조].
CRYSVITA 치료 시작 전에 공복 혈청 인산염 농도가 연령에 따른 참고 범위 미만이어야 합니다 [금기 사항 (4) 참조].
CRYSVITA는 피하 주사로 투여하며 의료 전문가가 투여해야 합니다.
CRYSVITA의 최대 주사량은 1.5mL입니다. 여러 번 주사해야 하는 경우 다른 주사 부위에 주사하십시오.
2.2 X-연관 저인산염혈증 소아 환자(6개월~18세 미만)
체중이 10kg 미만인 환자의 경우 권장 시작 용량은 체중 1kg당 1mg으로, 1mg 단위로 반올림하여 2주마다 투여합니다.
체중이 10kg 이상인 환자의 경우 권장 시작 용량 요법은 체중 1kg당 0.8mg으로, 10mg 단위로 반올림하여 2주마다 투여합니다. 최소 시작 용량은 10mg이며 최대 용량은 90mg입니다.
CRYSVITA 치료를 시작한 후 처음 3개월 동안은 매 4주마다 공복 혈청 인산염을 측정하고 그 후에는 적절히 측정합니다. 혈청 인산염이 연령에 따른 참고 범위의 하한 이상이고 5mg/dL 미만인 경우 동일한 용량으로 치료를 계속합니다. 연령에 따른 참고 범위 내에서 혈청 인산염을 유지하기 위해 아래의 용량 조정 일정을 따르십시오.
용량 조정
용량 조정 후 4주 후에 공복 혈청 인산염 수치를 재평가하십시오.
CRYSVITA는 4주마다 한 번 이상 조정하지 마십시오.
용량 증가:
체중이 10kg 미만인 환자의 경우 혈청 인산염이 연령에 따른 참고 범위 미만인 경우 용량을 1.5mg/kg으로 증가시킬 수 있으며, 1mg 단위로 반올림하여 2주마다 투여합니다. 추가 용량 증가가 필요한 경우 용량을 2mg/kg으로 증가시킬 수 있으며, 1mg 단위로 반올림하여 2주마다 투여합니다.
체중이 10kg 이상인 환자의 경우 혈청 인산염이 연령에 따른 참고 범위 미만인 경우 표 1에 표시된 용량 일정에 따라 2주마다 약 2mg/kg까지 단계적으로 용량을 증가시킬 수 있습니다(최대 용량 90mg).
| 체중 (kg) | 시작 용량 (mg) | 첫 번째 용량 증가 (mg) | 두 번째 용량 증가 (mg) |
|---|---|---|---|
| 10 – 14 | 10 | 15 | 20 |
| 15 – 18 | 10 | 20 | 30 |
| 19 – 31 | 20 | 30 | 40 |
| 32 – 43 | 30 | 40 | 60 |
| 44 – 56 | 40 | 60 | 80 |
| 57 – 68 | 50 | 70 | 90 |
| 69 – 80 | 60 | 90 | 90 |
| 81 – 93 | 70 | 90 | 90 |
| 94 – 105 | 80 | 90 | 90 |
| 106 이상 | 90 | 90 | 90 |
용량 감소:
혈청 인이 5mg/dL 이상인 경우, 다음 용량을 보류하고 4주 후에 혈청 인 수치를 재평가합니다. 환자는 CRYSVITA를 재시작하기 위해 연령에 따른 참조 범위 이하의 혈청 인을 가져야 합니다. 일단 혈청 인이 연령에 따른 참조 범위 이하로 떨어지면 치료를 재개할 수 있습니다.
체중이 10kg 미만인 환자의 경우, CRYSVITA를 0.5mg/kg의 체중으로 재시작하며, 가장 가까운 1mg로 반올림하여 2주마다 투여합니다. 체중이 10kg 이상인 환자의 경우, 표 2에 나와 있는 용량 일정에 따라 CRYSVITA를 재시작합니다.
| 이전 용량(mg) | 재시작 용량(mg) |
|---|---|
| 10 | 5 |
| 15 | 10 |
| 20 | 10 |
| 30 | 10 |
| 40 | 20 |
| 50 | 20 |
| 60 | 30 |
| 70 | 30 |
| 80 | 40 |
| 90 | 40 |
용량 감소 후, 용량 조정 후 4주 후에 혈청 인 수치를 재평가합니다. 재시작 용량 후에도 혈청 인 수치가 연령에 따른 참조 범위 이하인 경우, 용량 증가에 명시된대로 용량을 조정할 수 있습니다.
2.3 X-연관 저인산혈증을 가진 성인 환자(18세 이상)
성인에서의 권장 용량 요법은 1mg/kg의 체중이며, 가장 가까운 10mg로 반올림하여 최대 90mg이며, 4주마다 투여합니다.
CRYSVITA로 치료를 시작한 후, 치료의 첫 3개월 동안 투약 후 2주에 공복 혈청 인을 평가하고, 그 이후에는 적절하게 평가합니다. 혈청 인이 정상 범위 내에 있으면 동일한 용량으로 계속합니다.
용량 감소
용량 조정 후 2주 후에 공복 혈청 인 수치를 재평가합니다.
CRYSVITA를 4주마다보다 더 자주 조정하지 마십시오.
혈청 인이 정상 범위 이상인 경우, 다음 용량을 보류하고 4주 후에 혈청 인 수치를 재평가합니다. 환자는 CRYSVITA를 재시작할 수 있도록 혈청 인이 정상 범위 이하를 가져야 합니다. 일단 혈청 인이 정상 범위 이하로 떨어지면, 표 3에 나와 있는 용량 일정에 따라 최대 40mg까지 약 4주마다 초기 시작 용량의 약 절반으로 치료를 재시작할 수 있습니다. 용량의 모든 변경 후 2주에 혈청 인을 재평가합니다.
| 이전 용량(mg) | 재시작 용량(mg) |
|---|---|
| 40 | 20 |
| 50 | 20 |
| 60 | 30 |
| 70 | 30 |
| 80 및 그 이상 | 40 |
2.4 종양 유발성 골연화증 소아 환자 (2세에서 18세 미만)
소아의 권장 시작 용량은 체중 kg당 0.4mg을 2주마다 투여하며, 10mg 단위로 반올림하여 최대 용량은 2mg/kg (180mg 초과하지 않음)을 2주마다 투여합니다.
CRYSVITA 치료 시작 후, 처음 3개월 동안은 투여 후 2주째에 측정한 공복 혈청 인을 매월 평가하고, 그 이후에는 적절히 평가합니다. 혈청 인이 연령에 따른 참고 범위 내에 있으면 동일한 용량을 유지합니다. 연령에 따른 참고 범위 내에서 혈청 인을 유지하기 위해 아래 용량 조정 일정을 따르십시오.
용량 조정
용량 조정 후 4주 후에 공복 혈청 인 수치를 재평가합니다.
CRYSVITA는 4주마다보다 자주 조정하지 마십시오.
용량 증가
혈청 인이 연령에 따른 참고 범위 미만인 경우, 표 4에 따라 최대 용량인 2mg/kg을 2주마다 투여할 때까지 용량을 적정해야 합니다. 최대 용량은 180mg을 초과해서는 안 됩니다.
| 체중 (kg) |
시작 용량 (mg) |
첫 번째 용량 증가 (mg) |
두 번째 용량 증가 (mg) |
세 번째 용량* 증가 (mg) |
|---|---|---|---|---|
|
||||
| 10 – 14 | 5 | 10 | 15 | 20 |
| 15 – 18 | 5 | 10 | 20 | 25 |
| 19 – 31 | 10 | 20 | 25 | 30 |
| 32 – 43 | 10 | 30 | 40 | 50 |
| 44 – 56 | 20 | 40 | 50 | 70 |
| 57 – 68 | 20 | 50 | 70 | 90 |
| 69 – 80 | 30 | 60 | 80 | 100 |
| 81 – 93 | 30 | 70 | 100 | 120 |
| 94 – 105 | 40 | 80 | 110 | 140 |
| 106 이상 | 40 | 90 | 130 | 160 |
2.4 소아 환자(18세 미만)
CRYSVITA의 권장 시작 용량은 체중 kg당 0.5mg을 4주마다 투여하며, 소아의 경우 최대 용량은 180mg을 2주마다 투여합니다.
CRYSVITA 치료 시작 후, 처음 3개월 동안은 투여 후 2주째에 혈청 인을 매월 측정하고, 그 이후에는 적절히 측정합니다. 혈청 인이 정상 범위 내에 있으면 동일한 용량으로 계속 투여합니다. 혈청 인을 정상 범위 내로 유지하기 위해 아래 용량 조정 일정을 따르십시오.
용량 감소
혈청 인이 연령에 따른 참고 범위를 초과하는 경우, 다음 용량을 보류하고 4주 후에 혈청 인 수치를 재평가합니다. 환자는 CRYSVITA를 재개하기 위해 연령에 따른 참고 범위 미만의 혈청 인을 가져야 합니다. 혈청 인이 연령에 따른 참고 범위 미만이 되면, 소아의 경우 최대 180mg을 2주마다 투여하는 초기 시작 용량의 약 절반으로 치료를 재개할 수 있습니다. 용량 감소 후, 용량 조정 후 4주 후에 혈청 인 수치를 재평가합니다. 재개 용량 투여 후 연령에 따른 참고 범위 미만으로 유지되면, 표 4에 따라 용량을 조정할 수 있습니다.
용량 중단
환자가 기저 종양 치료(예: 수술적 절제 또는 방사선 치료)를 받는 경우, CRYSVITA 치료를 중단하고 치료가 완료된 후 혈청 인을 재평가해야 합니다. 혈청 인이 정상 하한 미만으로 유지되면, 환자의 초기 용량으로 CRYSVITA 용량을 재개해야 합니다. 연령에 따른 참고 범위 내로 혈청 인을 유지하기 위해 표 4에 따라 용량 조정을 수행하십시오.
2.5 종양 유발성 골연화증 성인 환자(18세 이상)
성인의 권장 시작 용량은 체중 kg당 0.5mg을 4주마다 투여하며, 최대 용량은 2mg/kg을 초과하지 않고 180mg을 2주마다 투여합니다.
CRYSVITA로 치료를 시작한 후, 처음 3개월 동안은 투여 후 2주째에 공복 혈청 인을 매월 측정하고, 그 이후에는 적절히 측정합니다. 혈청 인이 정상 범위 내에 있으면 동일한 용량으로 계속 투여합니다. 혈청 인을 정상 범위 내로 유지하기 위해 아래 용량 조정 일정을 따르십시오.
용량 조정
용량 조정 후 2주 후에 공복 혈청 인 수치를 재평가합니다.
CRYSVITA는 4주마다보다 자주 조정하지 마십시오.
용량 증가
혈청 인이 정상 범위 미만인 경우, 표 5에 따라 최대 용량인 2mg/kg을 초과하지 않고 180mg을 2주마다 투여할 때까지 용량을 조정해야 합니다. 정상 범위 하한보다 높은 혈청 인에 도달하지 못하는 개인의 경우, 의사는 4주마다 투여하는 총 용량을 나누어 2주마다 투여하는 것을 고려할 수 있습니다.
| 시작 용량 | 첫 번째 용량 증가‡ | 두 번째 용량 증가‡ | 세 번째 용량 증가‡ | 네 번째 용량 증가 | 다섯 번째 용량 증가 (최대 용량) |
|
|---|---|---|---|---|---|---|
| 용량 조정 후 2주 후 혈청 인이 정상 하한 미만인 경우 | 4주마다 0.5 mg/kg | 증가하여: 4주마다 1 mg/kg 또는 2주마다 0.5 mg/kg |
증가하여: 4주마다 1.5 mg/kg§ 또는 2주마다 0.75 mg/kg |
증가하여: 4주마다 2 mg/kg§ 또는 2주마다 1 mg/kg |
증가하여: 2주마다 180mg을 초과하지 않고 1.5 mg/kg |
증가하여: 2주마다 180mg을 초과하지 않고 2 mg/kg |
2.5 용량 조절
용량 감소
혈청 인 수치가 정상 범위를 초과하는 경우, 다음 투여를 중단하고 4주 후에 혈청 인 수치를 재평가합니다. 환자는 CRYSVITA를 재투여하기 위해 혈청 인 수치가 참고 범위 미만이어야 합니다. 혈청 인 수치가 참고 범위 미만이 되면, 성인의 경우 최대 180mg을 2주마다 투여하는 초기 시작 용량의 약 절반으로 치료를 재개할 수 있습니다. 용량 감소 후, 용량 조절 후 2주 후에 혈청 인 수치를 재평가합니다. 재투여 용량 후 수치가 참고 범위 미만으로 유지되면, 표 5에 따라 용량을 조절할 수 있습니다.
투여 중단
환자가 기저 종양 치료(예: 수술적 절제 또는 방사선 치료)를 받는 경우, CRYSVITA 치료를 중단하고 치료가 완료된 후 혈청 인을 재평가해야 합니다. 혈청 인이 정상 하한 미만으로 유지되면, 환자의 초기 용량으로 CRYSVITA 투여를 재개해야 합니다. 혈청 인을 참고 범위 내로 유지하기 위해 표 5에 따라 용량을 조절합니다.
2.6 누락된 용량
환자가 용량을 누락한 경우, 가능한 한 빨리 처방된 용량으로 CRYSVITA를 재개합니다. 누락된 용량을 방지하기 위해, 예정된 치료일 전후 3일 이내에 치료를 시행할 수 있습니다.
2.7 25-하이드록시 비타민 D 보충
25-하이드록시 비타민 D 수치를 모니터링합니다. 연령에 따른 정상 범위 내로 25-하이드록시 비타민 D 수치를 유지하기 위해 콜레칼시페롤 또는 에르고칼시페롤을 보충합니다. CRYSVITA 치료 중에 활성 비타민 D 유사체를 투여하지 마십시오 [금기 사항 (4) 참조].
2.8 피하 투여에 대한 일반적인 고려 사항
각 투여 시 주입 부위를 회전시켜 이전 주입과 다른 해부학적 위치(상완, 대퇴부 상단, 둔부 또는 복부의 모든 사분면)에 주입해야 합니다. 사마귀, 흉터 또는 피부가 압통, 멍, 붉은색, 단단하거나 손상된 부위에는 주입하지 마십시오. 주어진 투여일에 필요한 용량이 CRYSVITA 바이알 여러 개를 필요로 하는 경우, 두 바이알의 내용물을 결합하여 주입할 수 있습니다. 주입당 CRYSVITA의 최대 용량은 1.5mL입니다. 주어진 투여일에 여러 번 주입이 필요한 경우, 다른 주입 부위에 주입합니다. 반응 징후를 모니터링합니다 [경고 및 주의 사항 (5.3) 참조].
투여 전에 CRYSVITA를 시각적으로 검사하여 입자 및 변색이 있는지 확인합니다. CRYSVITA는 피하 주사용 무균, 방부제가 없는 투명하거나 약간 불투명하고 무색에서 옅은 갈색-황색 용액입니다. 용액이 변색되었거나 흐리거나 입자 또는 이물질이 포함되어 있는 경우 사용하지 마십시오.
3 제형 및 함량
주사제: 단회용 바이알에 담긴 맑거나 약간 유백색이며 무색에서 연한 갈색-황색의 용액, 10 mg/mL, 20 mg/mL 또는 30 mg/mL.
4 금기사항
CRYSVITA는 다음의 경우 금기입니다.
- 고인산혈증 위험 때문에 경구 인산염 및/또는 활성 비타민 D 유사체(예: calcitriol, paricalcitol, doxercalciferol, calcifediol)와 병용 [경고 및 주의사항 (5.2) 및 약물 상호작용 (7.1) 참조].
- 혈청 인 수치가 연령별 정상 범위 이내이거나 그 이상인 경우 [경고 및 주의사항 (5.2) 참조].
- 중증 신장애 또는 말기 신질환 환자의 경우 이러한 질환은 비정상적인 무기질 대사와 관련이 있기 때문입니다. [특정 집단에서의 사용 (8.6) 참조].
5 경고 및 주의사항
5.1 과민반응
CRYSVITA를 투여받은 환자에서 과민반응(예: 발진, 두드러기)이 보고되었습니다. 심각한 과민반응이 발생하면 CRYSVITA 투여를 중단하고 적절한 의학적 치료를 시작하십시오 [부작용 (6.1) 참조].
5.2 고인산혈증 및 신장석회화 위험
혈청 인 수치가 정상 상한치를 초과하는 증가는 신장석회화 위험 증가와 관련될 수 있습니다. 이미 CRYSVITA를 복용하고 있는 환자의 경우, 환자의 혈청 인 수치에 따라 투여 중단 및/또는 투여량 감소가 필요할 수 있습니다. 종양 유발성 골연화증이 있는 환자는 기저 종양 치료를 받는 동안 고인산혈증을 예방하기 위해 투여를 중단하고 조정해야 합니다 [투여 및 관리 (2) 및 부작용 (6.1) 참조].
5.3 주사 부위 반응
CRYSVITA 투여는 국소 주사 부위 반응을 유발할 수 있습니다. 심각한 주사 부위 반응이 발생하면 CRYSVITA 투여를 중단하고 적절한 의학적 치료를 시행하십시오 [부작용 (6.1) 참조].
6 부작용
다음의 유해 반응은 아래 및 라벨링의 다른 부분에 설명되어 있습니다.
6.1 임상 시험 경험
임상 시험은 매우 다양한 조건에서 수행되므로, 약물의 임상 시험에서 관찰된 유해 반응 발생률은 다른 약물의 임상 시험에서 관찰된 발생률과 직접 비교할 수 없으며, 실제로 관찰된 발생률을 반영하지 않을 수 있습니다.
XLH 소아 환자에서의 유해 반응
CRYSVITA는 3개의 소아 XLH 연구에서 연구되었습니다. 연구 1은 1세에서 12세 사이의 XLH 환자를 대상으로 한 무작위 배정, 공개 라벨 3상 연구로, CRYSVITA 치료 또는 경구 인산염 및 활성 비타민 D의 활성 대조군 치료를 무작위 배정했습니다(CRYSVITA N = 29, 활성 대조군 N = 32). 연구 2는 5세에서 12세 사이의 XLH 환자를 대상으로 한 공개 라벨 2상 연구입니다(N = 52). 연구 3은 1세에서 5세 미만의 XLH 환자를 대상으로 한 공개 라벨 2상 연구입니다(N = 13). 전반적으로 환자 인구는 1-12세(평균 연령 7.0세), 남성 49%, 백인 88%였습니다.
연구 1에서 CRYSVITA에 무작위 배정된 환자는 2주마다 약 0.90 mg/kg(범위 0.8-1.2 mg/kg)의 평균 용량을 투여받았습니다. 이 그룹과 활성 대조군 그룹의 모든 환자는 64주 동안 치료를 완료했습니다.
연구 1에서 64주 치료 기간 동안 CRYSVITA 그룹의 피험자 중 10% 이상에서 발생하고 활성 대조군 그룹의 피험자보다 발생률이 높은 유해 반응은 표 6에 나와 있습니다.
| 유해 반응 | CRYSVITA (N=29) n (%) |
활성 대조군 (N=32) n (%) |
|---|---|---|
| n = 이벤트가 발생한 환자 수; N = CRYSVITA 또는 활성 대조군을 적어도 1회 이상 투여받은 환자의 총 수 | ||
|
||
| 발열 | 16 (55) | 6 (19) |
| 주사 부위 반응* | 15 (52) | 0 (0) |
| 기침† | 15 (52) | 6 (19) |
| 구토 | 12 (41) | 8 (25) |
| 사지 통증 | 11 (38) | 10 (31) |
| 두통 | 10 (34) | 6 (19) |
| 치아 농양‡ | 10 (34) | 4 (13) |
| 충치 | 9 (31) | 2 (6) |
| 설사 | 7 (24) | 2 (6) |
| 비타민 D 감소§ | 7 (24) | 1 (3) |
| 변비 | 5 (17) | 0 (0) |
| 발진¶ | 4 (14) | 2 (6) |
| 메스꺼움 | 3 (10) | 1 (3) |
연구 2에서 26명의 환자는 64주차에 평균 1.05 mg/kg(범위 0.4 – 2.0 mg/kg)의 CRYSVITA를 2주마다 투여받았고, 나머지 26명의 환자는 4주마다 CRYSVITA를 투여받았습니다. 연구 2에서 평균 노출 기간은 124주였습니다. 연구 3에서 환자들은 40주차에 평균 0.90 mg/kg(범위 0.8-1.2 mg/kg)의 CRYSVITA를 2주마다 투여받았습니다. 연구 3에서 평균 노출 기간은 45주였습니다.
연구 2와 3에서 CRYSVITA를 투여받은 환자의 10% 이상에서 발생한 이상 반응은 표 7에 나와 있습니다.
| 이상 반응 | 연구 2 (N=52) n (%) |
연구 3 (N=13) n (%) |
전체 (N=65) n (%) |
|---|---|---|---|
| n = 사건이 발생한 환자 수; N = CRYSVITA를 최소 한 번 이상 투여받은 환자의 총 수 | |||
| 두통 | 38 (73) | 1 (8) | 39 (60) |
| 주사 부위 반응* | 35 (67) | 3 (23) | 38 (59) |
| 구토 | 25 (48) | 6 (46) | 31 (48) |
| 발열 | 23 (44) | 8 (62) | 31 (48) |
| 사지 통증 | 24 (46) | 3 (23) | 27 (42) |
| 비타민 D 감소† | 19 (37) | 2 (15) | 21 (32) |
| 발진‡ | 14 (27) | 1 (8) | 15 (23) |
| 치통 | 12 (23) | 2 (15) | 14 (22) |
| 근육통 | 9 (17) | 1 (8) | 10 (15) |
| 치아 농양 | 8 (15) | 3 (23) | 11 (17) |
| 현기증§ | 8 (15) | 0 (0) | 8 (12) |
과민 반응
연구 1(CRYSVITA군 N=29)에서 가장 흔한 과민 반응은 발진(10%), 주사 부위 발진(10%) 및 주사 부위 두드러기(7%)였습니다. 연구 2 및 3(N=65)에서 가장 흔한 과민 반응은 발진(22%), 주사 부위 발진(6%) 및 두드러기(5%)였습니다.
고인산혈증
소아 연구에서 고인산혈증 사례는 보고되지 않았습니다.
주사 부위 반응(ISR)
연구 1(CRYSVITA군 N=29)에서 환자의 52%가 CRYSVITA 주사 부위에 국소 주사 부위 반응(예: 주사 부위 두드러기, 홍반, 발진, 부종, 멍, 통증, 가려움증 및 혈종)을 보였습니다. 연구 2 및 3(N=65)에서 환자의 약 58%가 CRYSVITA 주사 부위에 국소 주사 부위 반응을 보였습니다. 주사 부위 반응은 일반적으로 중증도가 경미했으며, 주사 후 1일 이내에 발생하여 약 1~3일 지속되었으며, 치료가 필요하지 않았으며, 거의 모든 경우에 해소되었습니다.
XLH 성인 환자의 이상 반응
XLH 성인 환자에서 CRYSVITA의 안전성은 20~63세(평균 연령 41세)의 134명 환자를 대상으로 한 무작위 배정, 이중 맹검, 위약 대조 연구(연구 4)에서 입증되었습니다. 이 중 대부분은 백인/코카서스인(81%)이었고 여성(65%)이었습니다. 총 68명과 66명의 환자가 각각 CRYSVITA 또는 위약을 최소 1회 이상 투여받았습니다. CRYSVITA의 평균 투여량은 4주마다 피하 주사로 0.95mg/kg(범위 0.3~1.2mg/kg)이었습니다. 연구 4의 24주 위약 대조 기간 동안 CRYSVITA 치료를 받은 환자의 5% 이상에서 보고된 이상 반응과 위약군보다 2명 이상에서 보고된 이상 반응은 표 8에 나와 있습니다.
| 이상 반응 | CRYSVITA (N=68) n (%) |
위약 (N=66) n (%) |
|---|---|---|
| n = 사건이 발생한 환자 수; N = CRYSVITA 또는 위약을 최소 1회 이상 투여받은 환자의 총 수 | ||
| 요통 | 10 (15) | 6 (9) |
| 두통* | 9 (13) | 6 (9) |
| 치아 감염† | 9 (13) | 6 (9) |
| 불안한 다리 증후군 | 8 (12) | 5 (8) |
| 비타민 D 감소‡ | 8 (12) | 3 (5) |
| 현기증 | 7 (10) | 4 (6) |
| 근육 경련 | 5 (7) | 2 (3) |
| 변비 | 6 (9) | 0 (0) |
| 혈액 인산염 증가§ | 4 (6) | 0 (0) |
24주 위약 대조 연구는 모든 환자가 4주마다 CRYSVITA를 피하 주사로 투여받은 24주 오픈 라벨 치료 기간으로 이어졌습니다. 오픈 라벨 연장 기간 동안 새로운 유해 반응은 확인되지 않았습니다.
과민 반응
연구 4의 이중 맹검 기간 동안 CRYSVITA 및 위약 치료 그룹 모두에서 약 6%의 환자가 과민 반응을 경험했습니다. 이러한 사건은 경증 또는 중등도였으며 투약 중단을 요구하지 않았습니다.
고인산혈증
연구 4의 이중 맹검 기간 동안 CRYSVITA 치료 그룹의 7% 환자가 투약 감량 기준(단일 혈청 인 수치가 5.0mg/dL을 초과하거나 혈청 인 수치가 4.5mg/dL(정상 상한)을 두 번 초과)을 충족하는 고인산혈증을 경험했습니다. 고인산혈증은 투약 감량으로 관리되었습니다. 프로토콜 지정 기준을 충족하는 모든 환자의 투약량은 50% 감소했습니다. 지속적인 고인산혈증으로 인해 한 명의 환자만 두 번째 투약 감량이 필요했습니다.
주사 부위 반응(ISR)
연구 4의 이중 맹검 기간 동안 CRYSVITA 및 위약 치료 그룹 모두에서 약 12%의 환자가 주사 부위에 국소 반응(예: 주사 부위 반응, 홍반, 발진, 멍, 통증, 가려움증 및 혈종)을 보였습니다. 주사 부위 반응은 일반적으로 중증도가 경미했으며 주사 후 1일 이내에 발생하여 약 1~3일 지속되었으며 치료가 필요하지 않았으며 거의 모든 경우에 해결되었습니다.
불안한 다리 증후군(RLS)
연구 4의 이중 맹검 기간 동안 CRYSVITA 치료 그룹의 약 12%가 기저 불안한 다리 증후군(RLS)이 악화되었거나 경증에서 중등도의 새로운 RLS가 발생했습니다. 이러한 사건은 투약 중단으로 이어지지 않았습니다. 비중증 RLS는 다른 반복 투여 성인 XLH 연구에서도 보고되었습니다. 한 경우 기저 RLS가 악화되어 약물 투약 중단으로 이어졌고 이후 사건이 해결되었습니다.
척추 협착증
척추 협착증은 XLH 성인에게 흔하며 척수 압박이 보고되었습니다. XLH 성인에 대한 CRYSVITA 2상 및 3상 연구(총 N=176)에서 총 7명의 환자가 척추 수술을 받았습니다. 이러한 경우 대부분은 기존 척추 협착증의 진행과 관련된 것으로 보입니다. CRYSVITA 치료가 척추 협착증이나 척수 압박을 악화시키는지 여부는 알 수 없습니다.
TIO 환자의 유해 반응
TIO 환자에서 CRYSVITA의 안전성은 총 27명의 환자를 등록한 두 건의 단일군 임상 연구(연구 6 및 연구 7)에서 입증되었습니다. 14명의 환자가 남성이었으며 환자의 연령은 33세에서 73세까지였습니다. CRYSVITA의 평균 투약량은 4주마다 0.77mg/kg이었고 평균 노출 기간은 121주였습니다.
연구 6 및 연구 7의 통합 데이터에서 성인 TIO 환자에게 보고된 유해 반응은 표 9에 나와 있습니다.
| 유해 반응 | 전반적 (N=27) n (%) |
|---|---|
| 치아 농양* | 5 (19) |
| 근육 경련 | 5 (19) |
| 현기증 | 4 (15) |
| 변비 | 4 (15) |
| 주사 부위 반응† | 4 (15) |
| 발진‡ | 4 (15) |
| 두통 | 3 (11) |
| 비타민 D 결핍 | 2 (7) |
| 고인산혈증 | 2 (7) |
| 불안한 다리 증후군 | 2 (7) |
6.1 부작용
과민 반응
연구 6 및 7의 통합 데이터에서 환자의 22%가 과민 반응을 경험했습니다. 가장 흔한 과민 반응은 습진(11%)과 발진(11%)이었습니다. 이러한 사건은 중증도가 경미하거나 중간 정도였습니다.
고인산혈증
연구 6 및 7의 통합 데이터에서 2명의 환자(7%)가 고인산혈증을 경험했으며, 이는 용량 감소로 관리되었습니다.
주사 부위 반응
주사 부위 반응의 빈도는 15%(주사 부위 반응, 주사 부위 통증 및 주사 부위 부종)였습니다. 주사 부위 반응은 일반적으로 중증도가 경미했으며, 치료가 필요하지 않았고 모든 경우에 해결되었습니다.
불안한 다리 증후군
연구 6 및 7의 통합 데이터에서 2명의 환자(7%)가 불안한 다리 증후군의 증상을 경험했으며, 이는 경미했고 치료 중단이 필요하지 않았습니다.
6.2 면역원성
모든 치료용 단백질과 마찬가지로 면역원성이 발생할 가능성이 있습니다. 항체 형성의 검출은 검사의 민감도와 특이성에 크게 의존합니다. 또한, 검사에서 관찰된 항체(중화 항체 포함) 양성의 발생률은 검사 방법, 샘플 취급, 샘플 수집 시기, 동반 약물 및 기저 질환을 포함한 여러 요인의 영향을 받을 수 있습니다. 이러한 이유로, 아래에 설명된 연구에서 부로수맙-twza에 대한 항체 발생률을 다른 연구 또는 다른 제품의 항체 발생률과 비교하는 것은 오해의 소지가 있을 수 있습니다.
XLH 임상 연구에서 1~4세 환자는 0명(0/13), 5~12세 환자는 19%(10/52), 성인 환자는 15%(20/131)가 CRYSVITA를 투여받은 후 항약물 항체(ADA) 양성 판정을 받았습니다. 이 중 5~12세 환자 3명이 중화 항체 양성 판정을 받았습니다. ADA의 존재는 XLH 환자에서 부로수맙의 약동학, 약력학, 효능 및 안전성에 임상적으로 관련된 변화와 관련이 없었습니다.
TIO 임상 연구 한 건에서 성인 환자의 14%(2/14)가 CRYSVITA를 투여받은 후 ADA 양성 판정을 받았습니다. ADA 양성 환자 중 중화 항체 양성 판정을 받은 환자는 없었습니다. 또 다른 TIO 임상 연구에서 성인 환자 13명 중 ADA 양성 판정을 받은 환자는 없었습니다.
6.3 시판 후 경험
CRYSVITA의 시판 후 사용 중 다음과 같은 부작용이 확인되었습니다. 이러한 반응은 불확실한 규모의 모집단에서 자발적으로 보고되기 때문에, 항상 빈도를 신뢰할 수 있게 추정하거나 약물 노출과의 인과 관계를 확립할 수 있는 것은 아닙니다.
검사: CRYSVITA를 투여받은 소아 XLH 환자에서 혈액 인산염 증가가 보고되었습니다.
7 약물 상호작용
7.1 경구 인산염 및 활성 비타민 D 유사체
CRYSVITA와 경구 인산염 및/또는 활성 비타민 D 유사체를 병용하면 CRYSVITA 단독 투여 시 예상되는 것보다 인산염 농도가 더 높아집니다. 이러한 증가는 고인산혈증을 유발할 수 있으며, 이는 신장 석회화를 유발할 수 있습니다.
CRYSVITA와 경구 인산염 및/또는 활성 비타민 D 유사체의 병용은 금기입니다.
8 특정 집단에서의 사용
8.1 임신
위험 요약
CRYSVITA를 임산부에게 사용한 자료가 없어 약물 관련 유해 발달 결과 위험을 알 수 없습니다. 자궁 내에서 시노몰구스 원숭이에게 burosumab-twza를 노출시킨 결과 기형 유발 효과는 나타나지 않았습니다. 그러나 임신한 시노몰구스 원숭이에게 태아 사망 및 조산과 같은 유해 효과가 관찰되었지만, 이러한 효과는 최대 권장 인간 용량(MRHD) 2mg/kg을 2주마다 투여했을 때 인간 노출량의 15배(AUC 기준)에 해당하는 약물 노출에서 발생했으며, 모체 고인산혈증 및 태반 광물화를 동반했기 때문에 임상적 위험을 나타내는 것은 아닙니다(자료 참조 Data). 임신 기간 동안 혈청 인산염 수치를 모니터링해야 합니다([용량 및 투여(2.2) 참조 Dosage and Administration (2.2)]. Kyowa Kirin, Inc.의 유해 사례 보고 전화 1-844-768-3544로 임신 사실을 보고하십시오.
지정된 모집단의 주요 선천적 기형 및 유산의 배경 위험은 알 수 없습니다. 그러나 미국 일반 모집단의 주요 선천적 기형의 추정 배경 위험은 임상적으로 인지된 임신의 2%에서 4%이고, 유산의 위험은 15%에서 20%입니다.
자료
동물 자료
임신한 시노몰구스 원숭이를 대상으로 한 생식 독성 연구에서, burosumab-twza를 임신 20일부터 분만 또는 제왕절개(133일)까지 2주마다 1회 정맥 주사했습니다. 이 기간에는 기관 형성 기간이 포함됩니다. 투여 용량은 성인 MRHD 2mg/kg을 2주마다 투여했을 때 인간 노출량의 0.2배, 2배 및 15배였습니다. 치료 결과 태아 또는 자손에게 기형 유발 효과는 나타나지 않았습니다. 성인 MRHD 2mg/kg을 2주마다 투여했을 때 인간 노출량의 15배에서 태아 사망 증가, 임신 기간 단축 및 조산 발생률 증가가 관찰되었으며, 이는 모체 고인산혈증 및 태반 광물화와 동반되었습니다. burosumab-twza가 태아 혈청에서 검출되어 태반을 통한 이동을 나타냅니다. MRHD 2mg/kg을 2주마다 투여했을 때 인간 노출량의 15배에 노출된 어미의 태아 및 자손에게 고인산혈증이 나타났지만 이소성 광물화는 나타나지 않았습니다. burosumab-twza는 자손의 생존율을 포함한 출생 전후 성장에 영향을 미치지 않았습니다.
8.2 수유
위험 요약
모유에서 burosumab-twza의 존재 여부 또는 burosumab-twza가 모유 생산 또는 모유 수유 아기에게 미치는 영향에 대한 정보는 없습니다. 모체 IgG는 모유에 존재합니다. 그러나 모유 수유 아기의 국소 위장관 노출 및 제한적인 전신 노출에 대한 burosumab-twza의 영향은 알 수 없습니다. 수유 중 임상 데이터가 부족하여 수유 중 아기에게 CRYSVITA의 위험을 명확하게 판단할 수 없습니다. 따라서 모유 수유의 발달적 및 건강상 이점을 CRYSVITA에 대한 모체의 임상적 필요성과 CRYSVITA 또는 기저 모체 질환으로 인해 모유 수유 아기에게 발생할 수 있는 잠재적 유해 효과와 함께 고려해야 합니다.
8.4 소아 사용
CRYSVITA의 안전성 및 유효성은 6개월 이상 소아 환자에서 확립되었습니다. XLH가 있는 1세 이상 소아 환자의 안전성 및 유효성은 1~12세(연구 1)의 환자 61명을 대상으로 한 3상, 개방형, 활성 대조군 연구와 혈청 인산염 및 방사선학적 소견을 평가한 2개의 개방형 연구(5~12세 환자 52명(연구 2) 및 1~4세 환자 13명(연구 3))를 기반으로 합니다. 6개월에서 1세 사이 및 청소년 환자의 안전성 및 유효성은 1세에서 13세 미만의 XLH가 있는 소아 환자를 대상으로 한 연구에서 얻은 증거와 성인 및 소아 약동학(PK) 및 약력학(PD) 데이터의 추가 모델링 및 시뮬레이션을 통해 용량을 결정하는 데 도움이 됩니다([부작용(6.1) 및 임상 연구(14) 참조 Adverse Reactions (6.1) and Clinical Studies (14)].
6개월 미만의 XLH가 있는 소아 환자에서 CRYSVITA의 안전성 및 유효성은 확립되지 않았습니다.
TIO가 있는 2세 이상 소아 환자에서 CRYSVITA의 안전성 및 유효성은 TIO가 있는 성인 환자를 대상으로 한 연구에서 얻은 증거와 성인 및 소아 XLH 환자와 성인 TIO 환자의 PK 데이터의 추가 모델링 및 시뮬레이션을 통해 용량을 결정하는 데 도움이 됩니다.
2세 미만의 TIO가 있는 소아 환자에서 CRYSVITA의 안전성 및 유효성은 확립되지 않았습니다.
8.5 노인 사용
CRYSVITA의 임상 연구에는 65세 이상 환자가 충분히 포함되지 않아 젊은 환자와 반응이 다른지 여부를 확인할 수 없습니다. 다른 보고된 임상 경험에서 노인과 젊은 환자 간에 반응 차이가 확인되지 않았습니다. 일반적으로 노인 환자의 경우 용량 선택은 신중해야 하며, 간, 신장 또는 심장 기능 저하, 동반 질환 또는 다른 약물 치료의 빈도가 높기 때문에 일반적으로 용량 범위의 하한에서 시작해야 합니다.
8.6 신장애
신장애가 burosumab-twza의 약동학에 미치는 영향은 알 수 없습니다. 그러나 신장애는 비정상적인 무기질 대사를 유발하여 CRYSVITA 단독으로 예상되는 것보다 인산염 농도가 더 높아질 수 있습니다. 이러한 증가는 고인산혈증을 유발하여 신장 석회화를 유발할 수 있습니다.
CRYSVITA는 다음과 같이 정의된 중증 신장애 환자에게는 금기입니다.
- 추정 사구체 여과율(eGFR)이 15mL/min/1.73m2에서 29mL/min/1.73m2 사이이거나 말기 신장 질환(eGFR < 15mL/min/1.73m2)인 소아 환자
- 크레아티닌 청소율(CLcr)이 15mL/min에서 29mL/min 사이이거나 말기 신장 질환(CLcr < 15mL/min)인 성인 환자.
10 과다 복용
CRYSVITA 과다 복용에 대한 보고는 없습니다. CRYSVITA는 소아 임상 시험에서 최대 90mg까지 체중 kg당 2mg의 용량으로 2주마다 투여하여 용량 제한 독성 없이 투여되었습니다. XLH 성인 임상 시험에서 최대 1mg/kg 또는 4주마다 최대 총 용량 128mg까지 용량 제한 독성이 관찰되지 않았습니다. 비-XLH 토끼와 붉은털원숭이에서 생리적 혈청 인산염 수준을 초과하는 burosumab-twza 용량에서 여러 조직과 기관의 이소성 광화가 관찰되었습니다. 뼈 무기질 밀도, 뼈 광화 및 뼈 강도 감소를 포함한 뼈에 대한 부작용도 인간 노출보다 높은 노출에서 관찰되었습니다 [see Nonclinical Toxicology (13.2)].
과다 복용의 경우, 혈청 인산염 수치, 혈청 칼슘 수치 및 신장 기능을 즉시 측정하고 정상/기준 수치로 해결될 때까지 주기적으로 모니터링하는 것이 좋습니다. 고인산혈증의 경우, CRYSVITA를 중단하고 적절한 의학적 치료를 시작하십시오.
11 설명
부로수맙-트자는 중국 햄스터 난소 세포를 사용한 재조합 DNA 기술로 생산된 인간 면역글로불린 G 서브클래스 1(IgG1), 항인간 섬유아세포 성장 인자 23(FGF23) 항체입니다. 부로수맙-트자는 두 개의 중쇄(γ1-쇄) 분자와 두 개의 경쇄(κ-쇄) 분자로 구성됩니다. 각 중쇄는 아스파라긴 297(Asn297)에 N-결합 탄수화물 부분을 가지고 있습니다. 질량 분석법으로 결정된 부로수맙-트자의 분자량은 약 147,000입니다.
CRYSVITA(부로수맙-트자) 피하 주사용 주사제는 단회용 바이알에 무균, 방부제가 없는, 투명하거나 약간 유백색이며 무색에서 옅은 갈색-황색 용액으로 제공됩니다.
용액 1mL에는 부로수맙-트자 10mg, 20mg 또는 30mg, L-히스티딘(1.55mg), L-메티오닌(1.49mg), 폴리소르베이트 80(0.5mg), D-소르비톨(45.91mg)이 주사용수 USP에 함유되어 있습니다. pH를 6.25로 조절하기 위해 염산을 사용할 수 있습니다.
12 임상약리학
12.1 작용 기전
X-연관 저인산혈증은 신장 세뇨관 인산 재흡수와 1,25-디하이드록시 비타민 D의 신장 생산을 억제하는 과도한 섬유아세포 성장 인자 23(FGF23)에 의해 발생합니다. 부로수맙-트와자는 FGF23의 생물학적 활성에 결합하여 억제하여 신장 인산 재흡수를 회복시키고 혈청 1,25-디하이드록시 비타민 D 농도를 증가시킵니다.
12.2 약력학
XLH 및 TIO 환자에게 SC 투여 후, 더 높은 부로수맙-트와자 농도는 혈청 인 수치의 더 큰 증가와 관련이 있었습니다. 혈청 인의 증가는 가역적이었으며 전신 부로수맙-트와자의 제거와 함께 기준선으로 돌아왔습니다.
신장 세뇨관 최대 인산 재흡수율과 사구체 여과율의 비율(TmP/GFR)은 기준선에서 용량 의존적으로 증가했습니다 [임상 연구 (14) 참조].
부로수맙-트와자 치료 시작 후 혈청 총 FGF23의 상승이 관찰되었지만, 임상적 의미는 알려져 있지 않습니다.
12.3 약동학
다음 약동학적 매개변수는 별도로 명시되지 않는 한, 70kg 환자를 기준으로 승인된 권장 시작 용량을 투여받은 XLH 환자에서 관찰되었습니다. 모집단 PK 분석에 따르면 부로수맙-트와자의 PK 특성은 XLH 및 TIO 환자 간에 유사했습니다.
부로수맙-트와자는 0.1~1mg/kg(70kg XLH 환자를 기준으로 최대 승인 권장 용량의 0.08~0.8배)의 용량 범위 내에서 SC 주사 후 선형 약동학을 나타냈습니다.
부로수맙-트와자의 정상 상태 골 농도 평균(± SD)은 성인 XLH 환자에서 5.8(± 3.4) mcg/mL였습니다.
흡수
부로수맙-트와자의 평균 Tmax 값은 8~11일 범위였습니다.
분포
부로수맙-트와자의 명백한 분포 용적은 8L입니다.
제거
명백한 청소율은 0.290 L/day입니다. 부로수맙-트와자의 반감기는 약 19일입니다.
대사
부로수맙-트와자 대사의 정확한 경로는 규명되지 않았습니다. 부로수맙-트와자는 대사 경로를 통해 작은 펩타이드와 아미노산으로 분해될 것으로 예상됩니다.
특정 인구 집단
연령에 따라 부로수맙-트와자 약동학의 임상적으로 유의미한 차이는 관찰되지 않았습니다.
신장 또는 간 기능 장애가 부로수맙-트와자의 약동학에 미치는 영향은 알려져 있지 않습니다.
소아 환자
정상 상태 골 농도는 5~12세 XLH 환자에서 15.8(± 9.4) mcg/mL이고, 1~4세 XLH 환자에서 11.2(± 4.6) mcg/mL였습니다.
체중
부로수맙-트와자의 청소율과 분포 용적은 체중이 증가함에 따라 증가합니다.
약물 상호 작용 연구
CRYSVITA와 관련하여 약물 상호 작용 연구는 수행되지 않았습니다.
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 저해
장기 동물 연구에서 burosumab-twza의 발암 가능성은 평가되지 않았습니다.
burosumab-twza의 돌연변이 유발 가능성을 평가하기 위한 연구는 수행되지 않았습니다.
burosumab-twza의 영향을 평가하기 위한 동물에서의 특정 생식 연구는 수행되지 않았습니다.
최대 40주 동안 cynomolgus 원숭이에서 burosumab-twza를 사용한 독성학 연구에서 2주마다 2mg/kg의 최대 권장 인체 용량(MRHD)에서 인체 노출의 최대 16배에 해당하는 용량까지 여성 생식 기관에 대한 유의미한 부작용은 나타나지 않았습니다. 수컷 원숭이에서 2주마다 2mg/kg의 MRHD에서 인체 노출의 3~9배에 해당하는 용량에서 고인산혈증과 관련된 정세관 또는 정세관의 최소한의 광물화가 관찰되었지만, 정액 분석에서는 부작용이 나타나지 않았습니다.
13.2 동물 독성 및/또는 약리학
토끼와 cynomolgus 원숭이에서 burosumab-twza에 의한 FGF23 신호 전달 억제는 혈청 인산염과 1,25-디하이드록시 비타민 D를 증가시켰습니다. 생리적 혈청 인산염 수준을 초과하는 burosumab-twza 용량에서 여러 조직과 기관에서 이소성 광물화가 관찰되었습니다. XLH의 생쥐 모델인 야생형(WT) 및 저인산혈증 Hyp 생쥐에서 수행된 연구에서 이소성 광물화는 Hyp 생쥐에서 현저히 적었습니다.
성인 cynomolgus 원숭이에서 burosumab-twza는 2주마다 2mg/kg의 MRHD에서 인체 노출의 9~16배에 해당하는 용량에서 골 전환율, 무기질 함량 및/또는 무기질 밀도 및 피질 두께를 증가시켰습니다. 2주마다 2mg/kg의 MRHD에서 인체 노출의 9~11배에 해당하는 용량에서 성인 수컷 원숭이에서 골 무기질 밀도, 골 광물화 및 골 강도 감소를 포함한 골에 대한 부작용이 관찰되었습니다.
어린 cynomolgus 원숭이에서 burosumab-twza는 임상 소아 노출의 0.2~2배에 해당하는 용량에서 골 전환율, 무기질 함량 및/또는 무기질 밀도 및/또는 피질 두께를 증가시켰습니다. 소아 노출의 2배에 해당하는 용량에서 수컷 원숭이에서 골 광물화가 감소했지만 골 강도에는 영향을 미치지 않았습니다. burosumab-twza는 소아 노출의 최대 2배에 해당하는 용량에서 어린 원숭이의 골 발달에 영향을 미치지 않았습니다.
14 임상 연구
14.1 소아 X-연관 저인산혈증
CRYSVITA는 XLH 소아 환자 총 126명을 대상으로 한 세 가지 연구에서 평가되었습니다.
연구 1(NCT 02915705)은 1~12세의 소아 XLH 환자 61명을 대상으로 CRYSVITA 치료를 활성 대조군(경구 인산염 및 활성 비타민 D)과 비교한 64주 무작위, 공개 연구입니다. 첫 번째 용량 투여 시점에 환자의 평균 연령은 6.3세였고 44%는 남성이었습니다. 모든 환자는 기준선에서 RSS 점수 ≥ 2.0인 구루병의 방사선학적 증거를 보였으며 평균(SD) 4(3.1)년 동안 경구 인산염 및 활성 비타민 D 유사체를 투여받았습니다. 경구 인산염 및 활성 비타민 D 유사체는 연구 등록 전 7일간의 휴약 기간 동안 중단되었고 활성 대조군 환자에게 다시 시작되었습니다. 환자들은 2주마다 0.8mg/kg의 시작 용량으로 CRYSVITA를 투여받거나 경구 인산염(권장 용량 20-60mg/kg/일) 및 활성 비타민 D(권장 용량 칼시트리올 20-30ng/kg/일 또는 알파칼시돌 40-60ng/kg/일)를 투여받도록 무작위 배정되었습니다. 활성 대조군에 무작위 배정된 환자는 40주차에 약 41mg/kg/일(범위 18~110mg/kg/일)의 평균 경구 인산염 용량을, 64주차에 약 46mg/kg/일(범위 18mg/kg/일~166mg/kg/일)의 평균 경구 인산염 용량을 투여받았습니다. 또한 40주차에 평균 26ng/kg/일의 경구 칼시트리올 용량 또는 치료적으로 동등한 양의 알파칼시돌을, 64주차에 27ng/kg/일의 경구 칼시트리올 용량을 투여받았습니다. CRYSVITA군의 8명의 환자는 혈청 인 측정값을 기준으로 1.2mg/kg까지 증량했습니다. 모든 환자는 연구에서 최소 64주를 완료했습니다.
혈청 인
연구 1에서 CRYSVITA는 평균(SD) 혈청 인 수치를 기준선의 2.4(0.24)mg/dL에서 40주차에 3.3(0.43)mg/dL, 64주차에 3.3(0.42)mg/dL로 증가시켰습니다. 활성 대조군에서 평균(SD) 혈청 인 농도는 기준선의 2.3(0.26)mg/dL에서 40주차에 2.5(0.34)mg/dL로 증가했으며 64주차에 2.5(0.39)mg/dL로 기준선보다 낮게 유지되었습니다. TmP/GFR로 평가한 신장 인 재흡수 용량은 CRYSVITA 치료 환자에서 기준선의 평균(SD) 2.2(0.37)mg/dL에서 40주차와 64주차에 각각 3.4(0.67)mg/dL 및 3.3(0.65)mg/dL로 증가했습니다. 활성 대조군에서 평균(SD) TmP/GFR은 기준선의 2.0(0.33)mg/dL에서 40주차에 1.8(0.35)mg/dL로 감소했으며 64주차에 1.9(0.49)mg/dL로 기준선보다 낮게 유지되었습니다.
그림 1: 연구 1에서 1-12세 어린이의 치료군별 혈청 인 농도 및 기준선으로부터의 변화(mg/dL)(평균 ± SD)
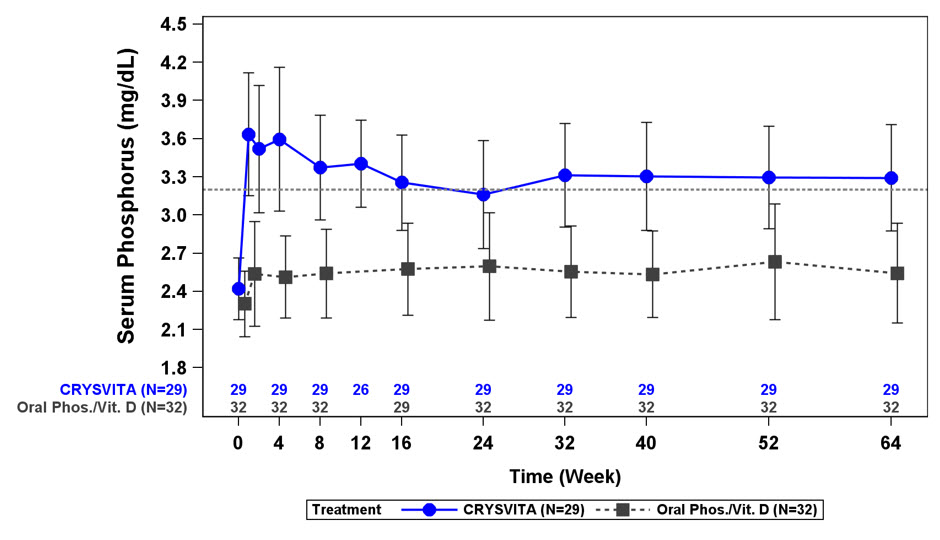
점선은 연구 1의 환자에 대한 정상 하한(3.2mg/dL)을 나타냅니다.
구루병의 방사선학적 평가
10점 Thacher Rickets Severity Score(RSS) 및 7점 Radiographic Global Impression of Change(RGI-C)를 사용하여 XLH 관련 구루병을 평가하기 위해 방사선 사진을 검사했습니다. RSS 점수는 단일 시점의 손목과 무릎 이미지를 기반으로 할당되며, 점수가 높을수록 구루병의 중증도가 더 높음을 나타냅니다. RGI-C 점수는 두 시점의 손목과 무릎 방사선 사진을 나란히 비교하여 할당되며, 점수가 높을수록 구루병의 방사선학적 증거가 더 많이 개선되었음을 나타냅니다. +2.0의 RGI-C 점수는 상당한 치유의 방사선학적 증거로 정의되었습니다.
연구 1에서 기준선 평균(SD) 총 RSS는 CRYSVITA군에서 3.2(0.98), 활성 대조군에서 3.2(1.14)였습니다. 40주 동안 CRYSVITA로 치료한 후 평균 총 RSS는 3.2에서 1.1(0.72)로 감소했고 활성 대조군에서는 3.2에서 2.5(1.09)로 감소했습니다. LS 평균(SE) RGI-C Global 점수는 40주차에 CRYSVITA군에서 +1.9(0.11), 활성 대조군에서 +0.8(0.11)였습니다(표 10 참조). 40주차에 CRYSVITA군의 29명 중 21명과 활성 대조군의 32명 중 2명이 +2.0 이상의 RGI-C Global 점수를 달성했습니다. 이러한 결과는 표 10과 같이 64주차에도 유지되었습니다.
| 평가 기준 시점 |
2주마다 CRYSVITA (N=29) |
활성 대조군 (N=32) |
|---|---|---|
| RSS 총점 | ||
| 기준선 평균(SD) | 3.2 (0.98) | 3.2 (1.14) |
| 총점에서 기준선으로부터의 LS 평균 변화*(감소는 개선을 나타냄) 및 95% CI | ||
| 40주차 | -2.0 (-2.33, -1.75) | -0.7 (-0.98, -0.43) |
| 64주차 | -2.2 (-2.46, -2.00) | -1.0 (-1.31, -0.72) |
| RGI-C Global 점수† | ||
| LS 평균 점수*(양수는 치유를 나타냄) 및 95% CI | ||
| 40주차 | +1.9 (+1.70, +2.14) | +0.8 (+0.56, +0.99) |
| 64주차 | +2.06 (+1.91, +2.20) | +1.03 (+0.77, +1.30) |
하지 골격 이상 (Lower Extremity Skeletal Abnormality)
연구 1에서, 하지 골격 이상은 서 있는 장골 방사선 사진에서 RGI-C로 평가되었습니다. 64주차에, CRYSVITA 그룹은 활성 대조군에 비해 더 큰 개선을 유지했습니다(LS 평균 [SE]: +1.25 [0.17] 대 +0.29 [0.12]; +0.97의 차이 (95% CI: +0.57, +1.37, GEE 모델)).
혈청 알칼리성 인산분해효소 활성 (Serum Alkaline Phosphatase Activity)
연구 1의 경우, 평균(SD) 혈청 총 알칼리성 인산분해효소 활성은 CRYSVITA 그룹에서 기준치 511(125) U/L에서 64주차에 337(86) U/L로 감소했습니다(평균 변화: -33%). 활성 대조군에서는 기준치 523(154) U/L에서 64주차에 495(182) U/L로 감소했습니다(평균 변화: -5%).
성장 (Growth)
연구 1에서, 64주 동안 CRYSVITA 치료는 서 있는 평균(SD) 키 Z 점수를 기준치 -2.32(1.17)에서 64주차에 -2.11(1.11)로 증가시켰습니다(LS 평균 변화(SE): +0.17(0.07)). 활성 대조군에서 평균(SD) 키 Z 점수는 기준치 -2.05(0.87)에서 64주차에 -2.03(0.83)으로 증가했습니다(LS 평균(SE) 변화: +0.02(0.04)). 64주차에 치료 그룹 간의 차이는 +0.14였습니다(95% CI: 0.00, +0.29).
연구 2(NCT 02163577)는 5세에서 12세 사이의 사춘기 이전 XLH 환자 52명을 대상으로 2주마다 CRYSVITA를 투여하는 것과 4주마다 투여하는 것을 비교한 무작위, 공개 라벨 연구입니다. 초기 16주 용량 적정 단계 후, 환자들은 48주 동안 2주마다 CRYSVITA 치료를 완료했습니다. 52명의 모든 환자가 연구에서 최소 64주를 완료했습니다. 어떤 환자도 중단하지 않았습니다. Burosumab-twza 용량은 투약 당일 공복 혈청 인 농도를 기준으로 공복 혈청 인 농도 3.5~5.0mg/dL을 목표로 조정되었습니다. 52명의 환자 중 26명은 최대 2mg/kg의 용량으로 2주마다 CRYSVITA를 투여받았습니다. 평균 용량은 16주차에 0.73mg/kg(범위: 0.3, 1.5), 40주차에 0.98mg/kg(범위: 0.4, 2.0), 60주차에 1.04mg/kg(범위: 0.4, 2.0)였습니다. 나머지 26명의 환자는 4주마다 CRYSVITA를 투여받았습니다. 연구 참여 시 환자의 평균 연령은 8.5세였고 46%는 남성이었습니다. 96%는 평균(SD) 7(2.4)년 동안 경구 인산염과 활성 비타민 D 유사체를 투여받았습니다. 경구 인산염과 활성 비타민 D 유사체는 연구 등록 전에 중단되었습니다. 환자의 94%는 기준치에 구루병의 방사선학적 증거를 보였습니다.
연구 3(NCT 02750618)은 1세에서 4세 사이의 소아 XLH 환자 13명을 대상으로 한 64주 공개 라벨 연구입니다. 환자들은 2주마다 0.8mg/kg의 용량으로 CRYSVITA를 투여받았으며, 3명의 환자는 혈청 인 측정값을 기준으로 1.2mg/kg까지 적정했습니다. 모든 환자가 연구에서 최소 40주를 완료했습니다. 어떤 환자도 중단하지 않았습니다. 연구 참여 시 환자의 평균 연령은 2.9세였고 69%는 남성이었습니다. 모든 환자는 기준치에 구루병의 방사선학적 증거를 보였고 12명의 환자는 평균(SD) 16.7(14.4)개월 동안 경구 인산염과 활성 비타민 D 유사체를 투여받았습니다. 경구 인산염과 활성 비타민 D 유사체는 연구 등록 전에 중단되었습니다.
혈청 인 (Serum Phosphorus)
연구 2에서, CRYSVITA는 2주마다 CRYSVITA를 투여받은 환자에서 평균(SD) 혈청 인 수치를 기준치 2.4(0.40)에서 40주차와 64주차에 3.3(0.40) 및 3.4(0.45)mg/dL로 증가시켰습니다. 사구체 여과율에 대한 인의 신세뇨관 최대 재흡수율 비율(TmP/GFR)은 이러한 환자에서 기준치 평균(SD) 2.2(0.49)에서 40주차와 64주차에 3.3(0.60) 및 3.4(0.53)mg/dL로 증가했습니다.
연구 3에서, CRYSVITA는 평균(SD) 혈청 인 수치를 기준치 2.5(0.28)mg/dL에서 40주차에 3.5(0.49)mg/dL로 증가시켰습니다.
구루병의 방사선학적 평가 (Radiographic Evaluation of Rickets)
연구 2에서, 2주마다 CRYSVITA를 투여받은 환자의 기준치 평균(SD) RSS 총점은 1.9(1.17)였습니다. 40주 동안 CRYSVITA 치료 후, 평균 총 RSS는 1.9에서 0.8로 감소했습니다(표 11 참조). 40주 동안 CRYSVITA 치료 후, 2주마다 CRYSVITA를 투여받은 환자의 평균 RGI-C 전체 점수는 +1.7이었습니다. 26명의 환자 중 18명이 +2.0 이상의 RGI-C 점수를 달성했습니다. 이러한 결과는 표 11에서 보는 바와 같이 64주차에 유지되었습니다.
연구 3에서, 13명의 환자의 기준치 평균(SD) 총 RSS는 2.9(1.37)였습니다. 40주 동안 CRYSVITA 치료 후, 평균 총 RSS는 2.9에서 1.2로 감소했으며 평균(SE) RGI-C 전체 점수는 +2.3(0.08)이었습니다(표 11 참조). 13명의 모든 환자가 +2.0 이상의 RGI-C 전체 점수를 달성했습니다.
| 평가변수 시점 (Endpoint Timepoint) |
2주마다 CRYSVITA (CRYSVITA Every 2 Weeks) | |
|---|---|---|
| 연구 2* (N=26) (Study 2 (N=26)) |
연구 3† (N=13) (Study 3 (N=13)) |
|
|
||
| RSS 총점 (RSS Total Score) | ||
| 기준치 평균 (SD) (Baseline Mean (SD)) | 1.9 (1.17) | 2.9 (1.37) |
| 총점에서 기준치 대비 LS 평균 변화(감소는 개선을 나타냄) 및 95% CI (LS Mean change from baseline in total score (reduction indicates improvement) with 95% CI) | ||
| 40주차 (Week 40) | -1.1 (-1.28, -0.85) | -1.7 (-2.03, -1.44) |
| 64주차 (Week 64) | -1.0 (-1.2, -0.79) | |
| RGI-C Global Score | ||
| LS 평균 점수(양수는 치유를 나타냄) 및 95% CI | ||
| Week 40 | +1.7 (+1.48, +1.84) | +2.3 (+2.16, +2.51) |
| Week 64 | +1.6 (+1.34, +1.78) | |
하지 골격 이상
연구 3에서, 서 있는 장각 방사선 사진을 사용하여 RGI-C로 평가한 하지 기형의 평균(SE) 변화는 40주차에 +1.3(0.14)였습니다.
혈청 알칼리성 인산분해효소 활성
연구 2의 경우, 2주마다 CRYSVITA를 투여받은 환자에서 평균(SD) 혈청 총 알칼리성 인산분해효소 활성은 기준치에서 462(110)U/L였으며 64주차에 354(73)U/L(-23%)로 감소했습니다.
연구 3의 경우, 평균(SD) 혈청 총 알칼리성 인산분해효소 활성은 기준치에서 549(194)U/L였으며 40주차에 335(88)U/L(평균 변화: -36%)로 감소했습니다.
성장
연구 2에서, 64주 동안 CRYSVITA 치료는 2주마다 CRYSVITA를 투여받은 환자에서 기준치 -1.72(1.03)에서 -1.54(1.13)로 서 있는 평균(SD) 신장 Z 점수를 증가시켰습니다(LS 평균 변화 +0.19(95% CI: 0.09~0.29)).
14.2 성인 X-연관 저인산혈증
연구 4(NCT 02526160)는 134명의 성인 XLH 환자를 대상으로 한 무작위, 이중맹검, 위약 대조 연구입니다. 이 연구는 24주간의 위약 대조 치료 단계와 그 후 모든 환자가 CRYSVITA를 투여받는 24주간의 공개 라벨 치료 기간으로 구성됩니다. CRYSVITA는 4주마다 1mg/kg 용량으로 투여되었습니다. 연구 참여 시 환자의 평균 연령은 40세(범위 19~66세)였으며 35%는 남성이었습니다. 모든 환자는 기준치에서 XLH/골연화증과 관련된 골격 통증이 있었습니다. 기준 평균(SD) 혈청 인 농도는 정상 하한보다 낮은 1.98(0.31)mg/dL였습니다. 연구 기간 동안 경구 인산염 및 활성 비타민 D 유사체는 허용되지 않았습니다. 연구에 등록된 134명의 환자 중 CRYSVITA 그룹의 한 환자는 24주간의 위약 대조 치료 기간 동안 치료를 중단했으며 7명의 환자는 공개 라벨 치료 기간 동안 CRYSVITA를 중단했습니다.
연구 5(NCT 02537431)는 장골능 골 생검의 조직학적 및 조직형태학적 평가에 의해 결정된 골연화증의 개선에 대한 CRYSVITA의 효과를 평가하기 위한 14명의 성인 XLH 환자를 대상으로 한 48주간의 공개 라벨, 단일군 연구입니다. 환자들은 4주마다 1mg/kg CRYSVITA를 투여받았습니다. 연구 참여 시 환자의 평균 연령은 40세(범위 25~52세)였으며 43%는 남성이었습니다. 연구 기간 동안 경구 인산염 및 활성 비타민 D 유사체는 허용되지 않았습니다.
혈청 인
연구 4에서 기준치의 평균(SD) 혈청 인은 위약군과 CRYSVITA군에서 각각 1.9(0.32) 및 2.0(0.30)mg/dL였습니다. 초기 24주간의 이중맹검, 위약 대조 기간 동안 투여 간격 중간점(투여 후 2주)의 평균(SD) 혈청 인은 위약군과 CRYSVITA군에서 각각 2.1(0.30) 및 3.2(0.53)mg/dL였으며 투여 간격 종료 시점의 평균(SD) 혈청 인은 위약군과 CRYSVITA군에서 각각 2.0(0.30) 및 2.7(0.45)mg/dL였습니다.
CRYSVITA로 치료받은 환자의 총 94%가 24주차까지 정상 하한(LLN) 이상의 혈청 인 수치에 도달한 반면 위약군에서는 8%에 불과했습니다(표 12 참조).
| 위약 (N = 66) |
CRYSVITA (N = 68) |
|
|---|---|---|
| 95% CI는 Wilson 점수 방법을 사용하여 계산됩니다. | ||
|
||
| 24주차까지 투여 간격 중간점에서 평균 혈청 인 > LLN 달성 – n (%) | 5 (8%) | 64 (94%) |
| 95% CI | (3.3, 16.5) | (85.8, 97.7) |
| p-값* | < 0.0001 | |
공개 라벨 치료 기간 동안 혈청 인은 지속적인 CRYSVITA 치료 동안 유지되었으며 48주차까지 효과 상실의 증거는 없었습니다.
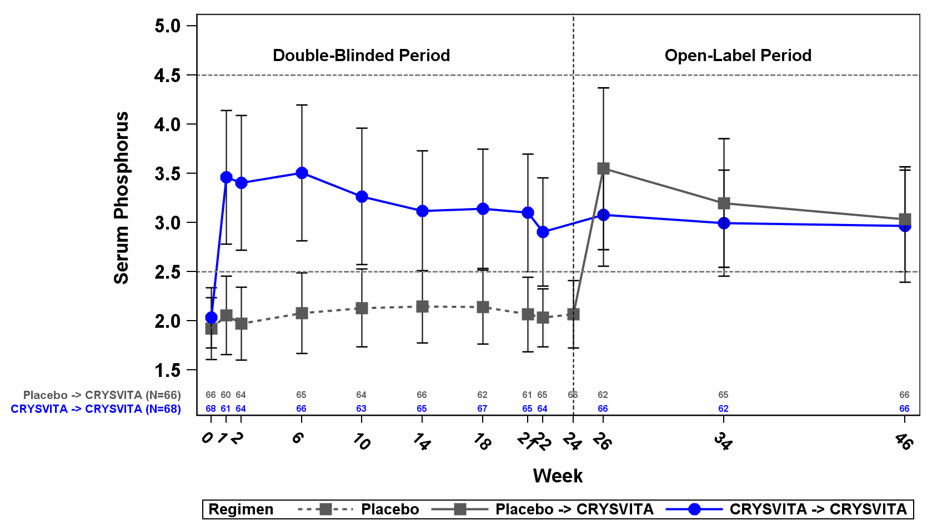
기준 시점에서 사구체 여과율 대비 신세뇨관 최대 인산 재흡수율(TmP/GFR)의 평균(SD) 비율은 위약군에서 1.60(0.37) mg/dL, CRYSVITA군에서 1.68(0.40) mg/dL이었습니다. 22주차(투여 간격의 중간 시점)에서 평균(SD) TmP/GFR은 위약군에서 1.69(0.37) mg/dL, CRYSVITA군에서 2.73(0.75) mg/dL이었습니다. 24주차(투여 간격의 종료 시점)에서 평균(SD) TmP/GFR은 위약군에서 1.73(0.42) mg/dL, CRYSVITA군에서 2.21(0.48) mg/dL이었습니다. 공개 라벨 치료 기간 동안 TmP/GFR은 48주차까지 CRYSVITA 치료를 지속하는 동안 안정적으로 유지되었습니다.
골연화증의 방사선학적 평가
연구 4에서 골연화증 관련 골절 및 가골절을 확인하기 위해 기준 시점에서 골격 검사를 수행했습니다. 골연화증 관련 골절은 외상 없이 두 피질골을 가로지르는 투과선으로 정의되며, 가골절은 외상 없이 하나의 피질골을 가로지르는 투과선으로 정의됩니다. 기준 시점에서 활성(미치유) 골절(12%) 또는 활성 가골절(47%) 중 하나를 가진 환자는 52%였습니다. 활성 골절 및 가골절은 주로 대퇴골, 경골/비골, 발의 중족골에 위치했습니다. 24주차에 이러한 활성 골절/가골절 부위를 평가한 결과, 표 13에서 보듯이 CRYSVITA군에서 위약군에 비해 완전 치유율이 더 높았습니다. 24주차까지 이중맹검, 위약 대조 치료 기간 동안 위약을 투여받은 66명의 환자에서 8건의 새로운 이상이 발생한 것에 비해 CRYSVITA를 투여받은 68명의 환자에서 총 6건의 새로운 골절 또는 가골절이 나타났습니다( 표 13 참조).
| 활성 골절 | 활성 가골절 | 총 골절 | ||||
|---|---|---|---|---|---|---|
| 위약 n (%) |
CRYSVITA n (%) |
위약 n (%) |
CRYSVITA n (%) |
위약 n (%) |
CRYSVITA n (%) |
|
| 기준 시점의 골절 수 | 13 | 14 | 78 | 51 | 91 | 65 |
| 24주차에 치유됨 | 0 (0%) | 7 (50%) | 7 (9%) | 21 (41%) | 7 (8%) | 28 (43%) |
공개표지 치료 기간 동안 CRYSVITA를 계속 투여받은 환자는 48주차에 골절의 지속적인 치유를 보였습니다[활성 골절(n = 8, 57%), 활성 유사 골절(n = 33, 65%)]. ‘위약에서 CRYSVITA로’ 그룹에서 48주차에 활성 골절(n = 6, 46%) 및 활성 유사 골절(n = 26, 33%)에 대한 골절 치유가 관찰되었습니다.
환자 보고 결과
연구 4에서는 환자가 보고한 XLH 관련 증상(통증, 관절 강직 및 신체 기능)을 평가했습니다.
24주차에 CRYSVITA군은 강직 중증도 점수(범위 0~100, 낮은 점수는 증상 개선을 반영함)에서 위약군(+0.3)에 비해 기준치(-7.9)에서 평균 개선을 보였습니다.
24주차에 환자가 보고한 통증 강도 또는 신체 기능 점수에서 CRYSVITA와 위약 간에 유의미한 차이는 나타나지 않았습니다.
뼈 조직 형태 측정
연구 5에서 48주 치료 후, 골기질 부피/뼈 부피(OV/BV)의 평균(SD) 점수가 기준치 26%(12.4)에서 11%(6.5)로 감소(-57% 변화)하여 10명의 환자에서 골연화증의 치유가 관찰되었습니다. 골기질 두께(O.Th)는 11명의 환자에서 평균(SD) 17(4.1)마이크로미터에서 12(3.1)마이크로미터로 감소(-33% 변화)했습니다. 무기질화 지연 시간(MLt)은 6명의 환자에서 평균(SD) 594(675)일에서 156(77)일로 감소하여 평균 변화는 -74%였습니다.
14.3 종양 유발 골연화증
CRYSVITA는 총 27명의 TIO 환자를 등록한 두 건의 연구에서 평가되었습니다.
연구 6(NCT 02304367)은 수술적 절제가 불가능하거나 위치를 찾을 수 없는 기저 종양으로 인한 FGF23 관련 저인산혈증 진단이 확정된 성인 환자 14명을 등록한 단일군 공개표지 연구입니다. 연구 6에 등록된 14명의 TIO 환자 중 8명은 남성이었고, 환자의 연령은 33세에서 68세(중앙값 59.5세)였습니다. 경구 인산염 및 활성 비타민 D 유사체는 연구 등록 2주 전에 중단되었습니다. 환자들은 4주마다 체중 기반 시작 용량 0.3mg/kg으로 CRYSVITA를 투여받았으며, 공복 혈청 인 수치가 2.5~4.0mg/dL에 도달하도록 용량을 조정했습니다. 평균 용량은 20주차에 0.83mg/kg, 48주차에 0.87mg/kg, 96주차에 0.77mg/kg, 144주차에 0.71mg/kg이었습니다.
연구 7(NCT 02722798)은 단일군 공개표지 연구입니다. 연구 7에서 TIO 진단이 확정된 성인 환자 13명이 CRYSVITA를 투여받았습니다. 연구 7에서 치료를 받은 13명의 TIO 환자 중 6명은 남성이었고, 환자의 연령은 41세에서 73세(중앙값 58.0세)였습니다. 경구 인산염 및 활성 비타민 D 유사체는 연구 등록 2주 전에 중단되었습니다. 환자들은 4주마다 체중 기반 시작 용량 0.3mg/kg으로 CRYSVITA를 투여받았으며, 공복 혈청 인 수치가 2.5~4.0mg/dL에 도달하도록 용량을 조정했습니다. 평균(SD) 용량은 48주차에 0.91(0.59)mg/kg, 88주차에 0.96(0.70)mg/kg이었습니다.
혈청 인
연구 6에서 CRYSVITA는 평균(SD) 혈청 인 수치를 기준치 1.60(0.47)mg/dL에서 24주차까지 용량 간격 중간점에서 평균 2.64(0.76)mg/dL로 증가시켰으며, 환자의 50%(7/14)가 24주차까지 용량 간격 중간점에서 평균 혈청 인 수치가 LLN 이상에 도달했습니다. 평균 혈청 인 농도의 증가는 144주차까지 LLN 근처 또는 이상으로 유지되었습니다(그림 3). 이러한 환자에서 인의 신세뇨관 최대 재흡수율 대 사구체 여과율의 비율(TmP/GFR)은 기준치 평균(SD) 1.12(0.54)mg/dL에서 48주차에 2.12(0.64)mg/dL로 증가했으며 144주차까지 안정적으로 유지되었습니다.
그림 3: 연구 6의 혈청 인 농도 및 기준치 대비 변화(mg/dL)
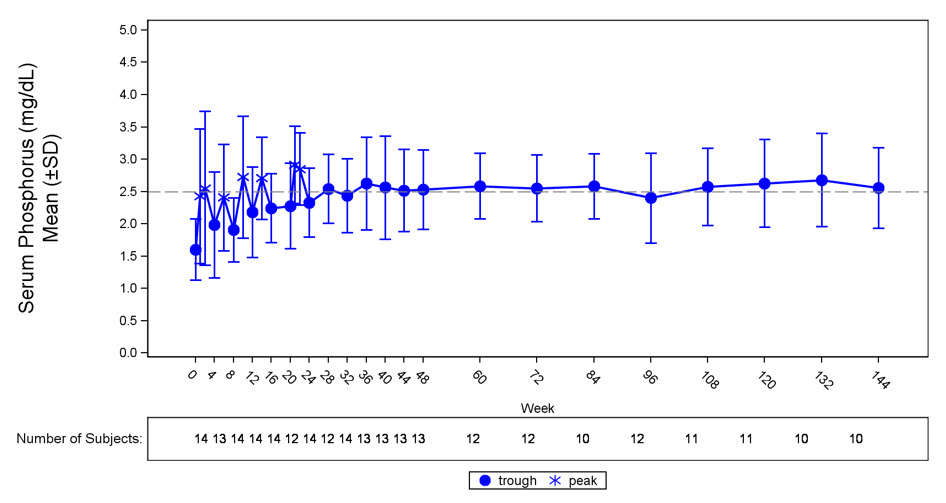
점선은 연구 6의 환자에 대한 정상 하한(2.5mg/dL)을 나타냅니다.
연구 7에서 CRYSVITA는 평균(SD) 혈청 인 수치를 기준치 1.62(0.49)mg/dL에서 24주차까지 용량 간격 중간점에서 평균 2.63(0.87)mg/dL로 증가시켰으며, 환자의 69%(9/13)가 용량 간격 중간점에서 평균 혈청 인 수치가 LLN 이상에 도달했습니다. 평균 혈청 인 농도는 88주차까지 LLN 이상으로 유지되었습니다. TmP/GFR로 평가한 신장 인 재흡수 용량은 기준치 평균(SD) 1.15(0.43)mg/dL에서 48주차에 2.30mg/dL(0.48)mg/dL로 증가했습니다.
뼈 조직 형태 측정
연구 6에서 짝을 이룬 뼈 생검을 받은 11명의 환자 중 9명에게서 기준치에 골연화증이 나타났으며, 48주 치료 후 치유를 평가했습니다. 기준치에 골연화증이 있는 이 9명의 환자에서 OV/BV는 기준치 평균(SD) 점수 21.2%(19.9)에서 13.9%(16.7)로 감소(-34% 변화)했습니다. O.Th는 평균(SD) 18.9(11.9)마이크로미터에서 12.1(10.1)마이크로미터로 감소(-36% 변화)했습니다. MLt는 3명의 환자에서 평균(SD) 667(414)일에서 331(396)일로 감소(-50% 변화)했습니다.
연구 7에서 짝을 이룬 뼈 생검을 받은 3명의 환자 모두에게서 기준치에 골연화증이 나타났으며, 48주 치료 후 치유를 평가했습니다. 이 3명의 환자에서 OV/BV는 기준치 평균(SD) 점수 14.0%(15.2)에서 9.2%(5.5)로 감소(-34% 변화)했습니다. O.Th는 평균(SD) 16.0(13.7)마이크로미터에서 13.5(7.1)마이크로미터로 감소(-16% 변화)했습니다.
골연화증의 방사선학적 평가
연구 6에서 14명의 환자 모두에게 기준치 및 연구 중 후속 시점에 99m테크네튬 표지 전신 뼈 스캔을 실시했습니다. 뼈 스캔을 통해 골연화증을 포함한 광범위한 뼈 질환에서 추적자 흡수 증가 부위를 평가할 수 있습니다. TIO 환자에서 뼈 스캔에서 추적자 흡수 증가는 비외상성 골절 및 유사 골절로 추정됩니다. 기준치에 모든 환자에게서 추적자 흡수 부위가 있었으며, 14명의 환자에서 총 249개의 뼈 이상이 있었습니다. 추적자 흡수 부위의 수는 48주차부터 144주차까지 감소했으며, 이는 뼈 이상의 치유를 시사합니다.
16 제공/보관 및 취급 방법
피하 주사용 CRYSVITA (burosumab-twza)는 멸균된 방부제가 없는 투명하거나 약간 유백색이며 무색에서 옅은 갈색-황색 용액으로 제공됩니다. 이 제품은 다음 농도로 상자당 단일 용량 바이알 1개로 제공됩니다.
10 mg/mL (NDC# 42747-102-01)
20 mg/mL (NDC# 42747-203-01)
30 mg/mL (NDC# 42747-304-01)
CRYSVITA 바이알은 사용 시까지 36°F~46°F(2°C~8°C)의 냉장 조건에서 원래 상자에 보관해야 합니다. 사용 시까지 CRYSVITA 바이알을 원래 상자에 보관하여 빛으로부터 보호하십시오.
CRYSVITA를 냉동하거나 흔들지 마십시오.
상자에 표시된 유효 기한이 지난 CRYSVITA는 사용하지 마십시오.
CRYSVITA 바이알은 단일 용량으로만 사용됩니다. 사용하지 않은 제품은 폐기하십시오.
17 환자 상담 정보
약물 상호작용 (Drug Interactions)
환자에게 경구 인산염 및/또는 활성 비타민 D 유사체 제품을 사용하지 않도록 조언하십시오. [see Contraindications (4)].
과민 반응 (Hypersensitivity Reactions)
CRYSVITA는 발진, 주사 부위 발진 및 두드러기와 같은 과민 반응을 유발할 수 있음을 환자에게 알리십시오. 이러한 반응이 발생하면 의사에게 연락하도록 환자에게 지시하십시오. [see Adverse Reactions (6.1)].
주사 부위 반응 (Injection Site Reactions)
CRYSVITA 주사 부위에 주사 부위 반응(예: 홍반, 발진, 부기, 멍, 통증, 가려움증, 두드러기 및 혈종)이 발생했음을 환자에게 알리십시오. 이러한 반응이 발생하면 의사에게 연락하도록 환자에게 지시하십시오. [see Adverse Reactions (6.1)].
하지불안 증후군 (Restless Legs Syndrome)
CRYSVITA는 RLS를 유발하거나 기존 RLS의 증상을 악화시킬 수 있음을 환자에게 알리십시오. 이러한 반응이 발생하면 의사에게 연락하도록 환자에게 지시하십시오. [see Adverse Reactions (6.1)].
임신 (Pregnancy)
임신 사실을 Kyowa Kirin, Inc. 부작용 보고 라인(1-844-768-3544)에 보고하십시오. [see Use in Specific Populations (8.1)].
Manufactured by:
Kyowa Kirin, Inc.
Princeton, NJ 08540
U.S. License No. 2077
주요 표시면 – 10 mg/mL 바이알
NDC 42747-102-01
CRYSViTA®
(burosumab-twza)
주사제
10 mg/mL
1 mL 단회용 바이알
쿄와 키린, Inc.
미국 허가 번호 2077

주요 표시 패널 – 10mg/mL 바이알 상자
NDC 42747-102-01
CRYSViTA®
(burosumab-twza)
주사제
10 mg/mL
피하 주사 전용
1회용 바이알
사용하지 않은 부분은 폐기하십시오
처방전에 의해서만 구입 가능
1 바이알
주요 표시면 – 20mg/mL 바이알
NDC 42747-203-01
CRYSViTA®
(burosumab-twza)
주사제
20 mg/mL
1 mL 단회용 바이알
쿄와 키린, Inc.
미국 허가 번호 2077

주요 표시 패널 – 20mg/mL 바이알 상자
NDC 42747-203-01
CRYSViTA®
(burosumab-twza)
주사제
20 mg/mL
피하 주사 전용
1회용 바이알
사용하지 않은 부분은 폐기하십시오
처방전에 의해서만 구입 가능
1 바이알
주요 표시면 – 30 mg/mL 바이알
NDC 42747-304-01
CRYSViTA®
(burosumab-twza)
주사제 (Injection)
30 mg/mL
1 mL 1회 용량 바이알 (1 mL single-dose vial)
Kyowa Kirin, Inc.
U.S. License No. 2077

주요 표시 패널 – 30mg/mL 바이알 상자
NDC 42747-304-01
CRYSViTA®
(burosumab-twza)
주사제
30 mg/mL
피하 주사 전용
1회용 바이알
사용하지 않은 부분은 폐기하십시오
처방전에 의해서만 구입 가능
1 바이알