
의약품 제조업체: Incyte Corporation (Updated: 2023-03-02)
처방 정보 하이라이트
JAKAFI® (룩솔리티닙) 정제, 경구용
미국 최초 승인: 2011
적응증 및 용법
용법 및 용량
안전성 및 유효성에 따라 용량을 개별화해야 합니다. 아래에 각 적응증에 대한 시작 용량이 명시되어 있습니다.
골수섬유증 (2.2)
- Jakafi의 시작 용량은 환자의 기준 혈소판 수에 따라 결정됩니다:
• 200 x 109/L 초과: 1일 2회 경구 투여 20mg
• 100 x 109/L ~ 200 x 109/L: 1일 2회 경구 투여 15mg
• 50 x 109/L ~ 100 x 109/L 미만: 1일 2회 경구 투여 5mg - 용량이 안정될 때까지 2~4주마다 완전 혈구 수를 모니터링하고, 그 후 임상적으로 필요한 경우 모니터링합니다. 혈소판 감소증의 경우 용량을 조절하거나 투여를 중단합니다.
진성적혈구증 (2.3)
- Jakafi의 시작 용량은 1일 2회 경구 투여 10mg입니다.
급성 이식편대숙주병 (2.4)
-
Jakafi의 시작 용량은 1일 2회 경구 투여 5mg입니다.
만성 이식편대숙주병 (2.5)
- Jakafi의 시작 용량은 1일 2회 경구 투여 10mg입니다.
제형 및 강도
정제: 5mg, 10mg, 15mg, 20mg 및 25mg. (3)
금기사항
없음. (4)
경고 및 주의사항
- 혈소판 감소증, 빈혈 및 호중구 감소증: 용량 감소 또는 중단 또는 수혈로 관리합니다. (5.1)
- 감염 위험: 감염의 징후 및 증상에 대해 환자를 평가하고 적절한 치료를 즉시 시작합니다. Jakafi 치료를 시작하기 전에 심각한 감염이 해결되어야 합니다. (5.2)
- 중단 또는 중지 후 증상 악화: 지지 요법으로 관리하고 Jakafi 치료 재개를 고려합니다. (5.3)
- 비흑색종 피부암 위험: 정기적인 피부 검사를 실시합니다. (5.4)
- 지질 상승: 치료 시작 후 8-12주에 지질 수치를 평가하고 필요에 따라 치료합니다. (5.5)
- 주요 심혈관계 이상 반응(MACE): MACE 발생 여부를 모니터링합니다. (5.6)
- 혈전증: 혈전증 증상을 평가하고 즉시 치료합니다. (5.7)
- 이차 악성 종양: 특히 현재 또는 과거 흡연자인 환자에서 이차 악성 종양 발생 여부를 모니터링합니다. (5.8)
이상 반응
- 골수섬유증 및 진성적혈구증에서 가장 흔한 혈액학적 이상 반응(발생률 > 20%)은 혈소판 감소증과 빈혈입니다. 가장 흔한 비혈액학적 이상 반응(발생률 ≥ 15%)은 멍, 어지러움, 두통 및 설사입니다. (6.1)
- 급성 이식편대숙주병에서 가장 흔한 혈액학적 이상 반응(발생률 > 50%)은 빈혈, 혈소판 감소증 및 호중구 감소증입니다. 가장 흔한 비혈액학적 이상 반응(발생률 > 50%)은 감염(병원체 불명)과 부종입니다. (6.1)
- 만성 이식편대숙주병에서 가장 흔한 혈액학적 이상 반응(발생률 > 35%)은 빈혈과 혈소판 감소증입니다. 가장 흔한 비혈액학적 이상 반응(발생률 ≥ 20%)은 감염(병원체 불명)과 바이러스 감염입니다. (6.1)
의심되는 이상 반응을 보고하려면 Incyte Corporation(1-855-463-3463) 또는 FDA(1-800-FDA-1088)에 연락하거나 www.fda.gov/medwatch를 방문하십시오.
약물 상호작용
특정 환자군에서의 사용
환자 상담 정보 및 FDA 승인 환자 라벨링은 17번을 참조하십시오.
개정: 2023년 3월
목차
FULL PRESCRIBING INFORMATION: CONTENTS*
1. 적응증 및 용법 (INDICATIONS AND USAGE)
1.1 골수섬유증 (Myelofibrosis)
1.2 진성적혈구증가증 (Polycythemia Vera)
1.3 급성 이식편대숙주병 (Acute Graft-Versus-Host Disease)
1.4 만성 이식편대숙주병 (Chronic
Graft-Versus-Host Disease)
2. 투여량 및 투여방법 (DOSAGE AND ADMINISTRATION)
2.1 안전성 평가를 위한 모니터링 (Monitoring to Assess Safety)
2.2 골수섬유증에 권장되는 투여량 (Recommended Dosage for Myelofibrosis)
2.3 진성적혈구증가증에 권장되는 투여량 (Recommended Dosage for Polycythemia Vera)
2.4 급성 이식편대숙주병에 권장되는 투여량 (Recommended Dosage for Acute Graft-Versus-Host Disease)
2.5 만성 이식편대숙주병에 권장되는 투여량 (Recommended Dosage for Chronic Graft-Versus-Host Disease)
2.6 강력한 CYP3A4 억제제 또는 Fluconazole 병용 투여 시 용량 조정 (Dose Modifications for Concomitant Use with Strong CYP3A4 Inhibitors or Fluconazole)
2.7 신장 또는 간 장애에 대한 용량 조정 (Dose Modifications for Renal or Hepatic Impairment)
2.8 투여 방법 (Method of Administration)
3. 제형 및 함량 (DOSAGE FORMS AND STRENGTHS)
4. 금기 (CONTRAINDICATIONS)
5. 경고 및 주의사항 (WARNINGS AND PRECAUTIONS)
5.1 혈소판감소증, 빈혈 및 호중구감소증 (Thrombocytopenia, Anemia and Neutropenia)
5.2 감염 위험 (Risk of Infection)
5.3 Jakafi 치료 중단 또는 중지 후 증상 악화 (Symptom Exacerbation Following Interruption or Discontinuation of Treatment with Jakafi)
5.4 비흑색종 피부암 (Non-Melanoma Skin Cancer (NMSC))
5.5
지질 상승 (Lipid Elevations)
5.6 주요 심혈관계 이상 사례 (Major Adverse Cardiovascular Events (MACE))
5.7 혈전증 (Thrombosis)
5.8 이차 악성종양 (Secondary Malignancies)
6. 이상반응 (ADVERSE REACTIONS)
6.1 임상시험 경험 (Clinical Trials Experience)
6.2 시판 후 경험 (Postmarketing Experience)
7. 약물 상호작용 (DRUG INTERACTIONS)
7.1 다른 약물이 Jakafi에 미치는 영향 (Effect of Other Drugs on Jakafi)
8. 특정 집단에서의 사용 (USE IN SPECIFIC POPULATIONS)
8.1 임신 (Pregnancy)
8.2 수유 (Lactation)
8.4 소아 사용 (Pediatric Use)
8.5 노인 사용 (Geriatric Use)
8.6 신장 장애 (Renal Impairment)
8.7 간 장애 (Hepatic Impairment)
10. 과다복용 (OVERDOSAGE)
11. 설명 (DESCRIPTION)
12. 임상 약리학 (CLINICAL PHARMACOLOGY)
12.1 작용 기전 (Mechanism of Action)
12.2 약력학 (Pharmacodynamics)
12.3 약동학 (Pharmacokinetics)
13. 비임상 독성학 (NONCLINICAL TOXICOLOGY)
13.1 발암성, 돌연변이 유발성, 생식력 손상 (Carcinogenesis, Mutagenesis, Impairment of Fertility)
14. 임상 연구 (CLINICAL STUDIES)
14.1 골수섬유증 (Myelofibrosis)
14.2 진성적혈구증가증 (Polycythemia Vera)
14.3
급성 이식편대숙주병 (Acute Graft-Versus-Host Disease)
14.4 만성 이식편대숙주병 (Chronic Graft-Versus-Host Disease)
16. 공급/보관 및 취급 방법 (HOW SUPPLIED/STORAGE AND HANDLING)
17. 환자 상담 정보 (PATIENT COUNSELING INFORMATION)
- *
- 전체 처방 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1. 적응증 및 용법
2. 복용량 및 투여 방법
2.1 안전성 평가를 위한 모니터링
Jakafi 치료 전:
- 전혈구 수 검사를 수행하십시오 [경고 및 주의사항 (5.1) 참조].
- 결핵, 단순 포진, 대상 포진, B형 간염을 포함한 과거 감염 이력에 대해 문의하십시오 [경고 및 주의사항 (5.2) 참조].
Jakafi 치료 중:
2.2 골수섬유증에 권장되는 용량
Jakafi의 권장 시작 용량은 혈소판 수에 따라 결정됩니다(표 1). 용량은 안전성과 효능에 따라 조절될 수 있습니다.
| 혈소판 수 | 시작 용량 |
| 200 x 109/L 초과 | 20 mg 경구, 1일 2회 |
| 100 x 109/L ~ 200 x 109/L | 15 mg 경구, 1일 2회 |
| 50 x 109/L ~ 100 x 109/L 미만 | 5 mg 경구, 1일 2회 |
혈소판 수 100 x 109/L 이상으로 치료를 시작하는 골수섬유증 환자의 혈액학적 독성에 대한 용량 조절 지침
치료 중단 및 재투여
혈소판 수가 50 x 109/L 미만이거나 절대 호중구 수(ANC)가 0.5 x 109/L 미만인 경우 치료를 중단하십시오.
혈소판 수가 50 x 109/L 초과이고 ANC가 0.75 x 109/L 초과로 회복된 후에 투여를 재개할 수 있습니다. 표 2는 이전에 중단한 후 Jakafi를 재투여할 때 사용할 수 있는 최대 허용 용량을 보여줍니다.
|
|
| 현재 혈소판 수 | Jakafi 치료 재개 시 최대 용량* |
| 125 x 109/L 이상 | 20 mg 1일 2회 |
| 100 ~ 125 x 109/L 미만 | 15 mg 1일 2회 |
| 75 ~ 100 x 109/L 미만 | 10 mg 1일 2회, 최소 2주 동안; 안정적인 경우 15 mg 1일 2회로 증량 가능 |
| 50 ~ 75 x 109/L 미만 | 5 mg 1일 2회, 최소 2주 동안; 안정적인 경우 10 mg 1일 2회로 증량 가능 |
| 50 x 109/L 미만 | 투약 중단 유지 |
ANC가 0.5 x 109/L 미만으로 치료가 중단된 후, ANC가 0.75 x 109/L 이상으로 회복되면 치료 중단 전 주의 최대 용량 미만으로 1일 1회 5mg 또는 1일 2회 5mg 중 더 높은 용량으로 다시 투여를 시작하십시오.
용량 감소
혈소판 감소증으로 인한 용량 중단을 피하기 위해 표 3에 요약된 대로 혈소판 수치가 감소하면 용량 감소를 고려해야 합니다.
| 혈소판 감소 시점의 용량 | |||||
| 혈소판 수치 | 25 mg 1일 2회 |
20 mg 1일 2회 |
15 mg 1일 2회 |
10 mg 1일 2회 |
5 mg 1일 2회 |
| 새로운 용량 |
새로운 용량 |
새로운 용량 |
새로운 용량 |
새로운 용량 |
|
| 100 ~ 125 x 109/L 미만 | 20 mg 1일 2회 |
15 mg 1일 2회 |
변경 없음 |
변경 없음 |
변경 없음 |
| 75 ~ 100 x 109/L 미만 | 10 mg 1일 2회 |
10 mg 1일 2회 |
10 mg 1일 2회 |
변경 없음 |
변경 없음 |
| 50 ~ 75 x 109/L 미만 | 5 mg 1일 2회 |
5 mg 1일 2회 |
5 mg 1일 2회 |
5 mg 1일 2회 |
변경 없음 |
| 50 x 109/L 미만 | 중단 | 중단 | 중단 | 중단 | 중단 |
혈소판 수가 100 x 109/L 이상인 골수섬유증 환자의 불충분한 반응에 따른 용량 조정
반응이 불충분하고 혈소판 및 호중구 수가 적절한 경우, 용량을 1일 2회 5 mg씩 최대 1일 2회 25 mg까지 증량할 수 있습니다. 치료 첫 4주 동안에는 용량을 증량해서는 안 되며, 2주마다 증량해서는 안 됩니다.
다음 조건을 모두 충족하는 환자의 용량 증량을 고려하십시오.
- 촉진 가능한 비장 길이의 치료 전 기준치에서 50% 감소 또는 컴퓨터 단층 촬영(CT) 또는 자기 공명 영상(MRI)으로 측정한 비장 부피의 35% 감소를 달성하지 못한 경우;
- 4주차에 혈소판 수가 125 x 109/L보다 크고 혈소판 수가 100 x 109/L 미만으로 떨어진 적이 없는 경우;
- ANC 수치가 0.75 x 109/L보다 큰 경우.
제한된 임상 데이터에 따르면 1일 2회 5 mg의 장기 유지 용량은 반응을 보이지 않았으며, 이 용량의 지속적인 사용은 이점이 잠재적 위험보다 큰 환자에게만 제한되어야 합니다. 6개월 치료 후 비장 크기 감소 또는 증상 개선이 없는 경우 Jakafi를 중단하십시오.
혈소판 수가 50 x 109/L에서 100 x 109/L 미만인 골수섬유증 환자의 혈액학적 독성에 대한 용량 조정
이 섹션은 Jakafi 치료 전 혈소판 수가 50 x 109/L에서 100 x 109/L 미만인 환자에게만 적용됩니다. Jakafi 치료 시작 전 혈소판 수가 100 x 109/L 이상인 환자의 혈액학적 독성에 대한 용량 조정은 2.2 (혈소판 수가 100 x 109/L 이상인 골수섬유증 환자의 혈액학적 독성에 대한 용량 조정 지침) 섹션을 참조하십시오.
치료 중단 및 재투여
혈소판 수가 25 x 109/L 미만이거나 ANC가 0.5 x 109/L 미만인 경우 치료를 중단하십시오.
혈소판 수가 35 x 109/L 이상으로 회복되고 ANC가 0.75 x 109/L 이상으로 회복된 후 투여를 재개할 수 있습니다. 혈소판 수가 25 x 109/L 미만으로 감소하거나 ANC가 0.5 x 109/L 미만으로 감소하여 용량 중단으로 이어지기 전 주의 최대 용량보다 낮은 1일 1회 5 mg 또는 1일 2회 5 mg 중 더 높은 용량으로 투여를 재개하십시오.
용량 감소
표 4에 설명된 대로 혈소판 수가 35 x 109/L 미만인 경우 Jakafi 용량을 줄이십시오.
| 혈소판 수 | 투여 권장 사항 |
| 25 x 109/L 미만 |
|
|
25 x 109/L에서 35 x 109/L 미만 그리고 이전 4주 동안 혈소판 수 감소가 |
|
|
25 x 109/L에서 35 x 109/L 미만 그리고 이전 4주 동안 혈소판 수 감소가 |
|
골수섬유증 환자 중 혈소판 수치가 50 x 109/L에서 100 x 109/L 미만인 환자에 대한 불충분한 반응에 따른 용량 조정
치료 첫 4주 동안 용량을 증량하지 마십시오. 또한 2주마다 용량을 증량하지 마십시오.
2.2절에 정의된 대로 반응이 불충분한 경우 (혈소판 수치가 100 x 109/L 이상인 골수섬유증으로 치료를 시작하는 경우 불충분한 반응에 따른 용량 조정 참조), 다음 조건을 충족하는 경우 용량을 5mg씩 1일 2회 최대 10mg까지 증량할 수 있습니다.
- 혈소판 수치가 최소 40 x 109/L로 유지되고,
- 지난 4주 동안 혈소판 수치가 20% 이상 감소하지 않았으며,
- ANC가 1 x 109/L 이상이고,
- 지난 4주 동안 이상반응 또는 혈액학적 독성으로 인해 용량을 줄이거나 중단하지 않았습니다.
6개월 이상 치료를 계속하는 것은 이점이 잠재적 위험보다 큰 환자에게만 국한되어야 합니다. 6개월 치료 후 비장 크기 감소 또는 증상 개선이 없는 경우 Jakafi를 중단하십시오.
2.3 진성적혈구증증에 권장되는 용량
Jakafi의 권장 시작 용량은 1일 2회 10mg입니다. 안전성과 효능에 따라 용량을 조정할 수 있습니다.
진성적혈구증증 환자에 대한 용량 조정 지침
용량 감소
표 5에 설명된 대로 헤모글로빈 및 혈소판 수치 감소에 대해 용량 감소를 고려해야 합니다.
| 헤모글로빈 및/또는 혈소판 수치 | 투여 권장 사항 |
| 헤모글로빈 12 g/dL 이상 및 혈소판 수치 100 x 109/L 이상 |
|
| 헤모글로빈 10~12 g/dL 미만 및 혈소판 수치 75~100 x 109/L 미만 |
|
| 헤모글로빈 8~10 g/dL 미만 또는 혈소판 수치 50~75 x 109/L 미만 |
|
| 헤모글로빈 8 g/dL 미만 또는 혈소판 수치 50 x 109/L 미만 |
|
치료 중단 및 재투여
헤모글로빈 수치가 8g/dL 미만, 혈소판 수치가 50 x 109/L 미만 또는 ANC가 1.0 x 109/L 미만인 경우 치료를 중단하십시오.
혈액학적 지표가 허용 가능한 수준으로 회복되면 투여를 재개할 수 있습니다.
표 6은 이전에 중단된 후 Jakafi를 재투여할 때 사용할 수 있는 용량을 보여줍니다.
표 6: 진성적혈구증가증: 혈액학적 지표에 대한 안전성 중단 후 Jakafi 재투여 용량
환자의 헤모글로빈, 혈소판 수치 또는 ANC 이상의 가장 심각한 범주를 사용하여 해당하는 최대 재투여 용량을 결정하십시오.
|
|
| 헤모글로빈, 혈소판 수치 또는 ANC | 최대 재투여 용량 |
| 헤모글로빈 8g/dL 미만 또는 혈소판 수치 50 x 109/L 미만 또는 ANC 1 x 109/L 미만 |
투약 중단 유지 |
| 헤모글로빈 8 ~ 10g/dL 미만 또는 혈소판 수치 50 ~ 75 x 109/L 미만 또는 ANC 1 ~ 1.5 x 109/L 미만 |
1일 2회 5mg* 또는 투약 중단을 초래한 용량보다 1일 2회 5mg 이하 |
| 헤모글로빈 10 ~ 12g/dL 미만 또는 혈소판 수치 75 ~ 100 x 109/L 미만 또는 ANC 1.5 ~ 2 x 109/L 미만 |
1일 2회 10mg* 또는 투약 중단을 초래한 용량보다 1일 2회 5mg 이하 |
| 헤모글로빈 12g/dL 이상 또는 혈소판 수치 100 x 109/L 이상 또는 ANC 2 x 109/L 이상 |
1일 2회 15mg* 또는 투약 중단을 초래한 용량보다 1일 2회 5mg 이하 |
헤모글로빈이 10g/dL 이상이고, 혈소판 수가 75 x 109/L 이상이며, ANC가 1.5 x 109/L 이상인 경우, 5mg 1일 2회 용량을 투여받는 동안 용량 중단이 필요했던 환자는 5mg 1일 2회 또는 5mg 1일 1회 용량으로 재투여할 수 있지만, 더 높은 용량으로는 재투여할 수 없습니다.
치료 재개 후 용량 관리
치료 중단 후 Jakafi를 재투여한 후 용량을 조정할 수 있지만, 최대 총 1일 용량은 용량 중단을 초래한 용량보다 5mg를 초과해서는 안 됩니다. 이에 대한 예외는 사혈 관련 빈혈 후 용량 중단이며, 이 경우 Jakafi 재투여 후 허용되는 최대 총 1일 용량은 제한되지 않습니다.
진성적혈구증가증 환자의 불충분한 반응에 따른 용량 조정
반응이 불충분하고 혈소판, 헤모글로빈 및 호중구 수가 적절한 경우, 용량을 5mg 1일 2회씩 최대 25mg 1일 2회까지 증량할 수 있습니다. 치료 첫 4주 동안 용량을 증량해서는 안 되며 2주마다 증량해서는 안 됩니다.
다음 조건을 모두 충족하는 환자의 용량 증량을 고려하십시오.
- 다음 중 하나 이상으로 입증된 불충분한 효능:
- 지속적인 사혈 필요
- 정상 범위 상한보다 높은 백혈구 수
- 정상 범위 상한보다 높은 혈소판 수
- 기준치에서 25% 미만으로 감소한 촉진 비장
- 혈소판 수 140 x 109/L 이상
- 헤모글로빈 12g/dL 이상
- ANC 1.5 x 109/L 이상
2.4 급성 이식편대숙주병에 대한 권장 용량
Jakafi의 권장 시작 용량은 5mg을 1일 2회 경구 투여하는 것입니다. Jakafi 투여 첫날 대비 ANC 및 혈소판 수가 50% 이상 감소하지 않은 경우 최소 3일 치료 후 용량을 10mg 1일 2회로 증량하는 것을 고려하십시오.
치료 용량의 코르티코스테로이드를 중단한 반응 환자의 경우 6개월 치료 후 Jakafi 감량을 고려하십시오. 약 8주마다 한 용량 수준씩 Jakafi를 감량하십시오(10mg 1일 2회에서 5mg 1일 2회, 5mg 1일 1회). Jakafi 감량 중 또는 감량 후 aGVHD 징후 또는 증상이 재발하는 경우 재치료를 고려하십시오.
급성 이식편대숙주병 환자에 대한 용량 조정 지침
치료 시작 전, 용량이 안정될 때까지 2~4주마다, 그리고 임상적으로 필요에 따라 혈소판 수와 ANC를 포함한 전체 혈구 수(CBC)와 빌리루빈을 모니터링하십시오.
표 7에 설명된 대로 이상반응에 대해 Jakafi의 용량을 조정하십시오. 용량 감량의 경우, 현재 Jakafi 10mg을 1일 2회 투여받는 환자는 용량을 5mg 1일 2회로 감량할 수 있습니다. 5mg을 1일 2회 투여받는 환자는 용량을 5mg 1일 1회로 감량할 수 있습니다. 5mg 1일 1회 용량의 Jakafi를 견딜 수 없는 환자는 임상적 및/또는 검사실적 지표가 회복될 때까지 치료를 중단해야 합니다.
| 검사실적 지표 | 투여 권장 사항 |
| 지지 요법 후 임상적으로 유의한 혈소판감소증 |
용량을 1단계 감량하십시오. |
| Jakafi와 관련된 것으로 간주되는 ANC 1 x 109/L 미만 |
최대 14일 동안 Jakafi 투여를 중단하십시오. 회복 시 1단계 낮은 용량으로 재개하십시오. |
| 총 빌리루빈 상승, 간 GVHD 없음 | 3.0−5.0 x ULN: 회복될 때까지 1단계 낮은 용량의 Jakafi를 계속 투여하십시오.
> 5.0−10.0 x ULN: 빌리루빈이 ≤ 1.5 x ULN이 될 때까지 최대 14일 동안 Jakafi 투여를 중단하십시오. 회복 시 현재 용량으로 재개하십시오. 총 빌리루빈 > 10.0 x ULN: 빌리루빈이 ≤ 1.5 x ULN이 될 때까지 최대 14일 동안 Jakafi 투여를 중단하십시오. 회복 시 1단계 낮은 용량으로 재개하십시오. |
| 총 빌리루빈 상승, 간 GVHD 있음 | > 3.0 × ULN: 회복될 때까지 1단계 낮은 용량의 Jakafi를 계속 투여하십시오. |
2.5 만성 이식편대숙주병에 권장되는 용량
Jakafi의 권장 시작 용량은 1일 2회 경구 복용하는 10mg입니다.
치료 용량의 코르티코스테로이드를 중단한 반응 환자의 경우 6개월 치료 후 Jakafi 감량을 고려하십시오. 약 8주마다 한 단계씩 Jakafi를 감량합니다(1일 2회 10mg에서 1일 2회 5mg, 1일 1회 5mg). Jakafi 감량 중 또는 감량 후 GVHD 징후 또는 증상이 재발하는 경우 재치료를 고려하십시오.
만성 이식편대숙주병 환자를 위한 용량 조정 지침
치료 시작 전, 용량이 안정될 때까지 2~4주마다, 그리고 임상적으로 필요에 따라 혈소판 수와 ANC를 포함한 전체 혈구 수(CBC) 및 빌리루빈을 모니터링하십시오.
표 8에 설명된 대로 부작용에 대해 Jakafi의 용량을 수정하십시오. 용량 감소의 경우, 현재 Jakafi 1일 2회 10mg을 복용하는 환자는 1일 2회 5mg으로 용량을 줄일 수 있습니다. 1일 2회 5mg을 복용하는 환자는 1일 1회 5mg으로 용량을 줄일 수 있습니다. 1일 1회 5mg 용량의 Jakafi를 견딜 수 없는 환자는 임상적 및/또는 실험실적 매개변수가 회복될 때까지 치료를 중단해야 합니다.
| 매개변수 | 투여 권장 사항 |
| 혈소판 수 20 × 109/L 미만 | Jakafi를 1단계 감량합니다. 7일 이내에 해결되면 초기 용량 수준으로 돌아갈 수 있습니다. 7일 이내에 해결되지 않으면 1단계 낮은 용량을 유지합니다. |
|
ANC 0.75 × 109/L 미만, Jakafi와 관련된 것으로 간주 |
Jakafi를 1단계 감량합니다. 회복되면 초기 용량 수준으로 재개합니다. |
|
ANC 0.5 × 109/L 미만, Jakafi와 관련된 것으로 간주 |
최대 14일 동안 Jakafi를 보류합니다. 회복되면 1단계 낮은 용량으로 재개합니다. ANC가 1.0 × 109/L보다 높으면 초기 용량 수준으로 재개할 수 있습니다. |
|
총 빌리루빈: 3.0-5.0 × ULN |
회복될 때까지 1단계 낮은 용량의 Jakafi를 계속합니다. 14일 이내에 해결되면 1단계 증량합니다. 14일 이내에 해결되지 않으면 감소된 용량 수준을 유지합니다. |
|
총 빌리루빈: > 5.0-10.0 × ULN |
해결될 때까지 최대 14일 동안 Jakafi를 보류합니다. 회복되면 현재 용량으로 재개합니다. 14일 이내에 해결되지 않으면 회복 시 1단계 낮은 용량으로 재개합니다. |
| 총 빌리루빈: > 10.0 × ULN |
해결될 때까지 최대 14일 동안 Jakafi를 보류합니다. 회복 시 1단계 낮은 용량으로 재개합니다. 14일 이내에 해결되지 않으면 중단합니다. |
| 기타 부작용: 3등급 | 회복될 때까지 1단계 낮은 용량의 Jakafi를 계속합니다. |
| 기타 부작용: 4등급 | Jakafi를 중단합니다. |
2.6 강력한 CYP3A4 억제제 또는 플루코나졸과의 병용 투여 시 용량 조정
표 9에 따라 강력한 CYP3A4 억제제 또는 200mg 이하 용량의 플루코나졸과 병용 투여하는 경우 Jakafi 용량을 조정하십시오. [약물 상호작용 (7)] Jakafi와 1일 200mg을 초과하는 플루코나졸 용량의 병용 투여는 피하십시오.
| 강력한 CYP3A4 억제제 또는 200mg 이하 용량의 플루코나졸과 병용 투여하는 환자의 경우 | 권장 Jakafi 용량 조정 |
| 혈소판 수가 다음과 같은 MF 환자의 시작 용량: | |
|
10mg 1일 2회 |
|
5mg 1일 1회 |
| PV 환자의 시작 용량: | 5mg 1일 2회 |
| MF 또는 PV 환자의 안정 용량 복용 시: | |
|
용량 50% 감소 (가장 가까운 사용 가능한 정제 강도로 반올림) |
|
5mg 1일 1회 |
|
강력한 CYP3A4 억제제 또는 플루코나졸 치료를 피하거나 강력한 CYP3A4 억제제 또는 플루코나졸 사용 기간 동안 Jakafi 치료를 중단 |
| aGVHD 또는 cGVHD 환자의 시작 용량: | |
| 200mg 이하 용량의 플루코나졸 |
aGVHD 환자의 경우 5mg 1일 1회; |
| 기타 CYP3A4 억제제 | 독성에 대해 혈구 수를 더 자주 모니터링하고 부작용이 발생하는 경우 Jakafi 용량을 조정하십시오. [용법 및 용량 (2.4, 2.5)]. |
2.7 신장 또는 간 장애에 대한 용량 조정
중등도에서 중증 신장애 또는 투석 중인 말기 신질환
표 10에 따라 중등도(CLcr 30~59mL/min)에서 중증(CLcr 15~29mL/min) 신장애 또는 투석 중인 말기 신질환(ESRD) 환자의 Jakafi 용량을 조정하십시오. 투석이 필요하지 않은 ESRD(CLcr 15mL/min 미만) 환자에게는 Jakafi 사용을 피하십시오. [특정 집단에서의 사용 참조(8.6)].
| ESRD = 말기 신질환, CLcr = 크레아티닌 청소율 | ||
| 신장애 상태 | 혈소판 수 | 권장 시작 용량 |
| MF 환자 |
||
|
중등도 또는 중증 |
150 x 109/L 초과 | 용량 조정 없음 |
| 100~150 x 109/L | 10mg 1일 2회 | |
| 50~100 x 109/L 미만 | 5mg 1일 1회 | |
| 50 x 109/L 미만 |
사용하지 마십시오 [특정 집단에서의 사용 참조 |
|
|
투석 중인 ESRD |
100~200 x 109/L | 투석 후 15mg 1회 |
|
200 x 109/L 초과 |
투석 후 20mg 1회 | |
| PV 환자 | ||
| 중등도 또는 중증 | 모두 | 5mg 1일 2회 |
| 투석 중인 ESRD | 모두 | 투석 후 10mg 1회 |
| aGVHD 환자 | ||
| 중등도 또는 중증 | 모두 | 5mg 1일 1회 |
| 투석 중인 ESRD | 모두 | 투석 후 5mg 1회 |
| cGVHD 환자 |
||
| 중등도 또는 중증 | 모두 | 5mg 1일 2회 |
| 투석 중인 ESRD | 모두 | 투석 후 10mg 1회 |
간 장애
표 11에 따라 간 장애 환자의 Jakafi 용량을 수정하십시오.
| 간 장애 상태 | 혈소판 수 | 권장 시작 용량 |
|
MF 환자 |
150 x 109/L 초과 | 용량 조정 없음 |
| 100 x 109/L ~ 150 x 109/L | 10 mg 1일 2회 | |
| 50 ~ 100 x 109/L 미만 | 5 mg 1일 1회 | |
| 50 x 109/L 미만 | 사용하지 마십시오 [특정 집단에서의 사용 참조(8.7)] | |
| PV 환자 경증, 중등도 또는 중증 (Child-Pugh Class A, B, C) |
모두 | 5 mg 1일 2회 |
| aGVHD 환자 | ||
|
간 GVHD가 없는 NCI 기준에 따른 경증, 중등도 또는 중증 |
모두 | 용량 조정 없음 |
|
1기, 2기 또는 3기 간 aGVHD |
모두 | 용량 조정 없음 |
|
4기 간 aGVHD |
모두 | 5 mg 1일 1회 |
| cGVHD 환자 |
||
|
간 GVHD가 없는 NCI 기준에 따른 경증, 중등도 또는 중증 |
모두 | 용량 조정 없음 |
|
1점 또는 2점 간 cGVHD |
모두 | 용량 조정 없음 |
|
3점 간 cGVHD |
모두 | 독성에 대해 혈구 수를 더 자주 모니터링하고 부작용이 발생하면 Jakafi 용량을 수정하십시오. [용량 및 투여 참조 (2.4, 2.5)]. |
2.8 투여 방법
Jakafi는 경구 투여하며 음식과 함께 또는 음식 없이 투여할 수 있습니다.
복용량을 놓친 경우 환자는 추가 용량을 복용해서는 안 되며 다음과 같이 처방된 일반 용량을 복용해야 합니다.
혈소판 감소증 이외의 이유로 Jakafi 치료를 중단할 때 Jakafi 용량을 점진적으로 줄이는 것을 고려할 수 있습니다(예: 매주 5mg씩 1일 2회).
정제를 삼킬 수 없는 환자의 경우 다음과 같이 비위관(8 French 이상)을 통해 Jakafi를 투여할 수 있습니다.
- 약 40mL의 물에 한 정제를 넣고 약 10분 동안 저어서 현탁합니다.
- 정제가 분산된 후 6시간 이내에 적절한 주사기를 사용하여 비위관을 통해 현탁액을 투여할 수 있습니다.
튜브를 약 75mL의 물로 헹궈야 합니다. 비위관을 통한 투여 중 경관 영양 제제가 Jakafi 노출에 미치는 영향은 평가되지 않았습니다.
3. 투여 형태 및 강도
5 mg 정제 – 한쪽 면에는 “INCY”, 다른 쪽 면에는 “5”가 새겨진 둥글고 흰색 정제.
10 mg 정제 – 한쪽 면에는 “INCY”, 다른 쪽 면에는 “10”이 새겨진 둥글고 흰색 정제.
15 mg 정제 – 한쪽 면에는 “INCY”, 다른 쪽 면에는 “15”가 새겨진 타원형 흰색 정제.
20 mg 정제 – 한쪽 면에는 “INCY”, 다른 쪽 면에는 “20”이 새겨진 캡슐 모양의 흰색 정제.
25 mg 정제 – 한쪽 면에는 “INCY”, 다른 쪽 면에는 “25”가 새겨진 타원형 흰색 정제.
4. 금기 사항
없음.
5. 경고 및 주의사항
5.1 혈소판 감소증, 빈혈 및 호중구 감소증
Jakafi 치료는 혈소판 감소증, 빈혈 및 호중구 감소증을 유발할 수 있습니다 [유해 반응 (6.1)].
용량을 줄이거나 Jakafi를 일시적으로 중단하여 혈소판 감소증을 관리합니다. 혈소판 수혈이 필요할 수 있습니다 [용법 및 용량 (2)].
빈혈이 발생하는 환자는 수혈 및/또는 Jakafi 용량 조절이 필요할 수 있습니다.
중증 호중구 감소증(ANC 0.5 × 109/L 미만)은 일반적으로 Jakafi를 중단하여 회복될 때까지 가역적이었습니다.
사전 치료 완전 혈구 수(CBC)를 수행하고 용량이 안정될 때까지 2~4주마다 CBC를 모니터링한 다음 임상적으로 필요한 경우 모니터링합니다 [용법 및 용량 (2)].
5.2 감염 위험
중증 세균성, 마이코박테리아성, 진균성 및 바이러스 감염이 발생했습니다 [유해 반응 (6.1)]. 활동성 중증 감염이 해결될 때까지 Jakafi 치료 시작을 지연합니다. Jakafi를 투여받는 환자의 감염 징후 및 증상을 관찰하고 신속하게 관리합니다. 임상 지침에 따라 적극적인 감시 및 예방적 항생제를 사용합니다.
결핵
Jakafi를 투여받는 환자에서 결핵 감염이 보고되었습니다. Jakafi를 투여받는 환자의 활동성 결핵의 징후와 증상을 관찰하고 신속하게 관리합니다.
Jakafi를 시작하기 전에 환자의 결핵 위험 요인을 평가해야 하며, 위험이 더 높은 환자는 잠복 감염 여부를 검사해야 합니다. 위험 요인에는 결핵 유병률이 높은 국가에 거주하거나 여행한 경력, 활동성 결핵 환자와의 밀접 접촉, 적절한 치료 과정을 확인할 수 없는 활동성 또는 잠복 결핵 병력 등이 포함되지만 이에 국한되지는 않습니다.
활동성 또는 잠복 결핵이 있는 환자의 경우 Jakafi를 시작하기 전에 결핵 치료 전문의와 상담하십시오. 활동성 결핵 치료 중 Jakafi를 계속 사용할지 여부는 전반적인 위험-이점 평가를 기반으로 결정해야 합니다.
진행성 다초점 백질뇌병증
진행성 다초점 백질뇌병증(PML)이 Jakafi 치료와 함께 발생했습니다. PML이 의심되는 경우 Jakafi를 중단하고 평가합니다.
대상포진 및 단순포진
Jakafi를 투여받는 환자에서 대상포진 감염이 보고되었습니다 [유해 반응 (6.1)]. 환자에게 대상포진의 초기 징후 및 증상에 대해 알리고 의심되는 경우 가능한 한 빨리 치료를 받도록 조언합니다.
Jakafi를 투여받는 환자에서 단순포진 바이러스 재활성화 및/또는 전파가 보고되었습니다 [유해 반응 (6.2)]. 단순포진 감염의 발생 여부를 모니터링합니다. 환자에게 단순포진 전파 증거가 나타나는 경우 Jakafi 치료 중단을 고려합니다. 환자는 임상 지침에 따라 신속하게 치료하고 모니터링해야 합니다.
B형 간염
알라닌 아미노전달효소 및 아스파르트산 아미노전달효소의 상승과 관련이 있거나 없는 B형 간염 바이러스 부하(HBV-DNA 역가) 증가가 Jakafi를 복용하는 만성 HBV 감염 환자에서 보고되었습니다. 만성 HBV 감염 환자에서 Jakafi의 바이러스 복제에 대한 영향은 알려져 있지 않습니다. 만성 HBV 감염 환자는 임상 지침에 따라 치료하고 모니터링해야 합니다.
5.3 Jakafi 치료 중단 또는 중지 후 증상 악화
Jakafi 중단 후 골수 증식성 신생물의 증상은 약 1주일 동안 치료 전 수준으로 돌아갈 수 있습니다. MF 환자 중 일부는 Jakafi 중단 후 발열, 호흡 곤란, 저혈압, DIC 또는 다기관 부전과 같은 하나 이상의 유해 사건을 경험했습니다. Jakafi 중단 후 또는 Jakafi 용량을 줄이는 동안 이러한 증상 중 하나 이상이 발생하는 경우, 간헐적 질병을 평가하고 치료하며 Jakafi의 재시작 또는 용량 증가를 고려합니다. 환자에게 의사와 상담하지 않고 Jakafi 치료를 중단하거나 중지하지 않도록 지시합니다. 혈소판 감소증 또는 호중구 감소증 이외의 이유로 Jakafi 치료를 중단하거나 중단하는 경우 [용법 및 용량 (2.8)], 갑자기 중단하는 것보다 Jakafi 용량을 점차적으로 줄이는 것을 고려합니다.
5.4 비흑색종 피부암(NMSC)
Jakafi로 치료받은 환자에서 기저 세포암, 편평 세포암 및 멀켈 세포암을 포함한 비흑색종 피부암이 발생했습니다. 정기적인 피부 검사를 실시합니다.
5.5 지질 수치 상승
Jakafi 치료는 총 콜레스테롤, 저밀도 지단백(LDL) 콜레스테롤 및 트리글리세라이드를 포함한 지질 매개변수 증가와 관련이 있습니다 [해당되는 이상 반응 (6.1)]. Jakafi로 치료받은 환자에서 이러한 지질 매개변수 상승의 심혈관 질환 발병률 및 사망률에 대한 영향은 밝혀지지 않았습니다. Jakafi 치료 시작 후 약 8-12주 후에 지질 매개변수를 평가하십시오. 고지혈증 관리에 대한 임상 지침에 따라 모니터링하고 치료하십시오.
5.6 주요 심혈관계 이상 반응 (MACE)
다른 JAK 억제제는 류마티스 관절염 환자에서 (TNF 억제제로 치료받은 환자와 비교하여) 심혈관 사망, 심근 경색 및 뇌졸중을 포함한 MACE 위험을 증가시켰습니다. Jakafi는 류마티스 관절염에 사용이 허가되지 않았습니다.
특히 현재 또는 과거 흡연자이고 다른 심혈관 위험 요인이 있는 환자의 경우 Jakafi 치료를 시작하거나 계속하기 전에 개별 환자의 이점과 위험을 고려하십시오. 환자는 심각한 심혈관계 이상 반응의 증상과 발생 시 취해야 할 조치에 대해 알고 있어야 합니다.
5.7 혈전증
다른 JAK 억제제는 류마티스 관절염 환자에서 (TNF 억제제로 치료받은 환자와 비교하여) 심부 정맥 혈전증(DVT), 폐색전증(PE) 및 동맥 혈전증을 포함한 혈전증 위험을 증가시켰습니다. Jakafi는 류마티스 관절염에 사용이 허가되지 않았습니다. 임상 시험에서 Jakafi로 치료받은 MF 및 PV 환자의 경우 혈전색전증 발생률은 Jakafi 및 대조군 치료 환자에서 유사했습니다.
혈전증 증상이 있는 환자는 즉시 적절하게 평가하고 치료해야 합니다.
5.8 이차 악성 종양
다른 JAK 억제제는 류마티스 관절염 환자에서 (TNF 억제제로 치료받은 환자와 비교하여) 비흑색종 피부암(NMSC)을 제외한 림프종 및 기타 악성 종양의 위험을 증가시켰습니다. Jakafi는 류마티스 관절염에 사용이 허가되지 않았습니다. 현재 또는 과거 흡연자는 추가적인 위험 증가가 있습니다.
특히 알려진 이차 악성 종양(성공적으로 치료된 NMSC 제외)이 있는 환자, 악성 종양이 발생한 환자 및 현재 또는 과거 흡연자의 경우 Jakafi 치료를 시작하거나 계속하기 전에 개별 환자의 이점과 위험을 고려하십시오.
6. 부작용
다음과 같은 임상적으로 중요한 이상 반응은 이 약의 설명서 다른 부분에서 자세히 설명하고 있습니다.
- 혈소판 감소증, 빈혈 및 호중구 감소증 [경고 및 주의 사항 (5.1)] 참조]
- 감염 위험 [경고 및 주의 사항 (5.2)] 참조]
- Jakafi 치료 중단 또는 중지 후 증상 악화 [경고 및 주의 사항 (5.3)] 참조]
- 비흑색종 피부암 [경고 및 주의 사항 (5.4)] 참조]
- 지질 상승 [경고 및 주의 사항 (5.5)] 참조]
- 중대한 심혈관계 이상 반응 (MACE) [경고 및 주의 사항 (5.6)] 참조]
- 혈전증 [경고 및 주의 사항 (5.7)] 참조]
- 이차 악성 종양 [경고 및 주의 사항 (5.8)] 참조]
6.1 임상 시험 경험
임상 시험은 매우 다양한 조건 하에서 수행되므로, 특정 약물의 임상 시험에서 관찰된 이상 반응 발생률을 다른 약물의 임상 시험에서 관찰된 발생률과 직접 비교할 수 없으며, 실제 발생률을 반영하지 않을 수도 있습니다.
골수섬유증
Jakafi의 안전성은 6건의 임상 연구에서 617명의 환자를 대상으로 평가되었으며, 중앙 추적 기간은 10.9개월이었고, 여기에는 2건의 3상 연구에서 MF 환자 301명이 포함되었습니다.
이 두 건의 3상 연구에서 환자의 Jakafi 노출 중앙 기간은 9.5개월(범위 0.5~17개월)이었으며, 89%의 환자가 6개월 이상, 25%의 환자가 12개월 이상 치료를 받았습니다. 1일 2회 15mg으로 치료를 시작한 환자는 111명, 1일 2회 20mg으로 치료를 시작한 환자는 190명이었습니다. 1일 2회 15mg(치료 전 혈소판 수 100~200 x 109/L) 및 1일 2회 20mg(치료 전 혈소판 수 200 x 109/L 초과)으로 치료를 시작한 환자 중 각각 65%와 25%가 치료 첫 8주 이내에 시작 용량보다 낮은 용량 감소가 필요했습니다.
Jakafi의 이중 눈가림, 무작위 배정, 위약 대조 연구에서 Jakafi로 치료받은 155명의 환자 중 가장 흔한 이상 반응은 혈소판 감소증과 빈혈이었습니다 [표 13 참조]. 혈소판 감소증, 빈혈 및 호중구 감소증은 용량 의존적 효과입니다. 가장 흔한 비혈액학적 이상 반응 세 가지는 멍, 어지러움 및 두통이었습니다 [표 12 참조].
원인과 관계없이 이상 사례로 인한 치료 중단은 Jakafi로 치료받은 환자의 11%와 위약으로 치료받은 환자의 11%에서 관찰되었습니다.
표 12는 무작위 치료 기간 동안 이중 눈가림, 위약 대조 연구에서 Jakafi를 투여받은 환자에게서 발생한 가장 흔한 비혈액학적 이상 반응을 보여줍니다.
| Jakafi (N=155) |
위약 (N=151) |
|||||
| 이상 반응 | 모든 등급* (%) |
3등급 (%) |
4등급 (%) |
모든 등급 (%) |
3등급 (%) |
4등급 (%) |
| Bruising† | 23 | < 1 | 0 | 15 | 0 | 0 |
| Dizziness‡ | 18 | < 1 | 0 | 7 | 0 | 0 |
| Headache | 15 | 0 | 0 | 5 | 0 | 0 |
| Urinary Tract Infections§ | 9 | 0 | 0 | 5 | < 1 | < 1 |
| Weight Gain¶ | 7 | < 1 | 0 | 1 | < 1 | 0 |
| Flatulence | 5 | 0 | 0 | < 1 | 0 | 0 |
| Herpes Zoster# | 2 | 0 | 0 | < 1 | 0 | 0 |
주요 이상 반응 설명
빈혈
두 건의 3상 임상 연구에서, CTCAE 2등급 이상 빈혈의 첫 발현까지의 중앙값 기간은 약 6주였습니다. 빈혈로 인해 치료를 중단한 환자는 1명 미만(<1%)이었습니다. Jakafi를 투여받은 환자에서, 헤모글로빈의 평균 감소는 치료 8~12주 후 기준치보다 약 1.5~2.0 g/dL 감소하는 최저치에 도달한 후 점차 회복되어 기준치보다 약 1.0 g/dL 낮은 새로운 안정 상태에 도달했습니다. 이러한 양상은 치료 중 수혈을 받았는지 여부에 관계없이 모든 환자에서 관찰되었습니다.
무작위 대조 연구에서, Jakafi로 치료받은 환자의 60%와 위약을 투여받은 환자의 38%가 무작위 치료 기간 동안 적혈구 수혈을 받았습니다. 수혈을 받은 환자 중, Jakafi로 치료받은 환자의 월별 수혈 단위 중앙값은 1.2였고, 위약으로 치료받은 환자는 1.7이었습니다.
혈소판 감소증
두 건의 3상 임상 연구에서, 3등급 또는 4등급 혈소판 감소증이 발생한 환자의 경우, 발현까지의 중앙값 기간은 약 8주였습니다. 혈소판 감소증은 일반적으로 용량 감소 또는 용량 중단으로 회복되었습니다. 혈소판 수치가 50 × 109/L 이상으로 회복될 때까지의 중앙값 기간은 14일이었습니다. Jakafi를 투여받은 환자의 5%와 대조군 치료를 받은 환자의 4%에게 혈소판 수혈이 시행되었습니다. 혈소판 감소증으로 인해 치료를 중단한 환자는 Jakafi를 투여받은 환자의 <1%와 대조군 치료를 받은 환자의 <1%였습니다. Jakafi 투여를 시작하기 전 혈소판 수치가 100 × 109/L~200 × 109/L인 환자는 혈소판 수치가 200 × 109/L보다 높은 환자에 비해 3등급 또는 4등급 혈소판 감소증의 빈도가 더 높았습니다(17% 대 7%).
호중구 감소증
두 건의 3상 임상 연구에서, 호중구 감소증으로 인해 Jakafi의 용량을 줄이거나 투여를 중단한 환자는 1%였습니다.
표 13은 위약 대조 연구에서 Jakafi 또는 위약으로 치료받은 환자에 대해 보고된 임상 혈액학 이상의 빈도와 중증도를 보여줍니다.
| Jakafi (N=155) |
위약 (N=151) |
|||||
|
검사 항목 |
모든 등급† (%) |
3등급 (%) |
4등급 (%) |
모든 등급 (%) |
3등급 (%) |
4등급 (%) |
| 혈소판 감소증 | 70 | 9 | 4 | 31 | 1 | 0 |
| 빈혈 | 96 | 34 | 11 | 87 | 16 | 3 |
| 호중구 감소증 | 19 | 5 | 2 | 4 | <1 | 1 |
위약 대조 연구의 추가 데이터
- Jakafi로 치료받은 환자의 25%와 위약으로 치료받은 환자의 7%에서 알라닌 아미노전이효소(ALT)의 새롭게 발생하거나 악화된 1등급 이상의 이상이 나타났습니다. 2등급 이상 상승의 발생률은 Jakafi의 경우 2%였으며, 3등급은 1%, 4등급 ALT 상승은 없었습니다.
- Jakafi로 치료받은 환자의 17%와 위약으로 치료받은 환자의 6%에서 아스파르트 아미노전이효소(AST)의 새롭게 발생하거나 악화된 1등급 이상의 이상이 나타났습니다. Jakafi의 경우 2등급 AST 상승의 발생률은 < 1%였으며, 3등급 또는 4등급 AST 상승은 없었습니다.
- Jakafi로 치료받은 환자의 17%와 위약으로 치료받은 환자의 < 1%에서 콜레스테롤의 새롭게 발생하거나 악화된 1등급 상승이 나타났습니다. Jakafi의 경우 2등급 콜레스테롤 상승의 발생률은 < 1%였으며, 3등급 또는 4등급 콜레스테롤 상승은 없었습니다.
진성적혈구증가증
무작위 배정, 공개, 활성 대조 연구에서, 하이드록시우레아에 내성이 있거나 내약성이 없는 PV 환자 110명은 Jakafi를, 111명은 최상의 이용 가능한 치료법을 받았습니다 [임상 연구 (14.2)]를 참조하십시오. 가장 흔한 이상 반응은 빈혈이었습니다. 원인과 관계없이 이상 사례로 인한 치료 중단은 Jakafi로 치료받은 환자의 4%에서 관찰되었습니다. 표 14는 무작위 치료의 32주차까지 발생한 가장 흔한 비혈액학적 이상 반응을 보여줍니다.
| Jakafi (N=110) |
최상의 이용 가능한 치료법 (N=111) |
|||
|
이상 반응 |
모든 등급* (%) |
3-4등급 (%) |
모든 등급 (%) |
3-4등급 (%) |
| 설사 | 15 | 0 | 7 | < 1 |
| 현기증† | 15 | 0 | 13 | 0 |
| 호흡곤란‡ | 13 | 3 | 4 | 0 |
| 근육 경련 | 12 | < 1 | 5 | 0 |
| 변비 | 8 | 0 | 3 | 0 |
| 대상포진§ | 6 | < 1 | 0 | 0 |
| 오심 | 6 | 0 | 4 | 0 |
| 체중 증가¶ | 6 | 0 | < 1 | 0 |
| 요로감염# | 6 | 0 | 3 | 0 |
| 고혈압 | 5 | < 1 | 3 | < 1 |
임상적으로 중요한 실험실 이상은 표 15에 나와 있습니다.
| Jakafi (N=110) |
최상의 이용 가능한 치료법 (N=111) |
|||||
| 실험실 매개변수 | 모든 등급† (%) |
3등급 (%) |
4등급 (%) |
모든 등급 (%) |
3등급 (%) |
4등급 (%) |
| 혈액학 | ||||||
| 빈혈 | 72 | < 1 | < 1 | 58 | 0 | 0 |
| 혈소판 감소증 | 27 | 5 | < 1 | 24 | 3 | < 1 |
| 호중구 감소증 | 3 | 0 | < 1 | 10 | < 1 | 0 |
| 화학 | ||||||
| 고콜레스테롤혈증 | 35 | 0 | 0 | 8 | 0 | 0 |
| ALT 상승 | 25 | < 1 | 0 | 16 | 0 | 0 |
| AST 상승 | 23 | 0 | 0 | 23 | < 1 | 0 |
| 고중성지방혈증 | 15 | 0 | 0 | 13 | 0 | 0 |
급성 이식편대숙주병
단일군, 공개표지 연구에서, 71명의 성인(18-73세)이 스테로이드를 단독 또는 다른 면역억제제와 병용하여 치료받지 못한 aGVHD에 대해 Jakafi로 치료받았습니다 [참조 임상 연구(14.3)]. Jakafi 치료의 중앙값 기간은 46일(범위, 4~382일)이었습니다.
Jakafi에 대한 치명적인 유해 반응은 없었습니다. 치료 중단으로 이어진 유해 반응은 환자의 31%에서 발생했습니다. 치료 중단으로 이어진 가장 흔한 유해 반응은 감염(10%)이었습니다. 표 16은 검사실 이상을 제외한 유해 반응을 보여줍니다.
| Jakafi (N=71) | ||
| 유해 반응* | 모든 등급† (%) |
3-4등급 (%) |
|
감염 (병원체 명시되지 않음) |
55 |
41 |
| 부종 | 51 | 13 |
| 출혈 | 49 | 20 |
| 피로 | 37 | 14 |
| 세균 감염 | 32 | 28 |
| 호흡곤란 | 32 | 7 |
| 바이러스 감염 | 31 | 14 |
| 혈전증 | 25 | 11 |
| 설사 | 24 | 7 |
| 발진 | 23 | 3 |
| 두통 | 21 | 4 |
| 고혈압 | 20 | 13 |
| 현기증 | 16 | 0 |
Jakafi 치료 중 관찰된 선택적 실험실 이상은 표 17에 제시되어 있습니다.
|
||
| Jakafi (N=71) | ||
| 치료 중 최악 등급 | ||
| 실험실 매개변수 | 모든 등급* (%) |
3-4등급 |
| 혈액학 | ||
| 빈혈 | 75 | 45 |
| 혈소판 감소증 | 75 | 61 |
| 호중구 감소증 | 58 | 40 |
| 화학 | ||
| ALT 상승 | 48 | 8 |
| AST 상승 | 48 | 6 |
| 고중성지방혈증 | 11 | 1 |
만성 이식편대숙주병
3상 무작위배정, 공개, 다기관 연구에서, 165명의 환자는 Jakafi로 치료받았고 158명의 환자는 스테로이드를 사용한 치료 또는 다른 면역억제제를 사용한 치료에 실패한 cGVHD에 대해 최상의 이용 가능한 치료를 받았습니다 [임상 연구(14.4)]; 65명의 환자가 최상의 이용 가능한 치료에서 Jakafi 치료로 전환되어 총 230명의 환자가 Jakafi로 치료받았습니다. 연구에서 Jakafi 노출의 중앙값 기간은 Jakafi군에서 49.7주(범위, 0.7~144.9주)였습니다. 109명(47%)의 환자가 1년 이상 Jakafi를 복용했습니다.
Jakafi와 관련된 5건의 치명적인 이상 반응이 있었는데, 여기에는 독성 표피괴사용해증 1건과 호중구감소증, 빈혈 및/또는 혈소판감소증 4건이 포함됩니다. Jakafi로 치료받은 환자의 18%에서 치료 중단으로 이어지는 이상 반응이 발생했습니다. 27%에서 용량 조절로 이어지는 이상 반응이 발생했고, 23%에서 치료 중단으로 이어지는 이상 반응이 발생했습니다. 가장 흔한 혈액학적 이상 반응(발생률 > 35%)은 빈혈과 혈소판감소증입니다. 가장 흔한 비혈액학적 이상 반응(발생률 ≥ 20%)은 감염(병원체 불명)과 바이러스 감염입니다.
표 18은 무작위 치료의 7주차 1일차까지 발생한 가장 빈번한 비실험실적 이상 반응을 보여줍니다.
| 이상 반응* |
Jakafi (N = 165) |
최상의 이용 가능한 치료 (N = 158) |
||
|
모든 등급† |
3등급 이상 |
모든 등급 |
3등급 이상 |
|
| 감염 및 기생충 감염 |
||||
| 감염 (병원체 불명) | 45 | 15 | 44 | 16 |
| 바이러스 감염 | 28 | 5 | 23 | 5 |
| 근골격계 및 결합조직 장애 |
||||
| 근골격계 통증 | 18 | 1 | 13 | 0 |
| 일반 장애 및 투여 부위 상태 | ||||
| 발열 | 16 | 2 | 9 | 1 |
| 피로 | 13 | 1 | 10 | 2 |
| 부종 | 10 | 1 | 12 | 1 |
| 혈관 장애 |
||||
| 고혈압 | 16 | 5 | 13 | 7 |
| 출혈 | 12 | 2 | 15 | 2 |
| 호흡기, 흉부 및 종격동 장애 | ||||
| 기침 | 13 | 0 | 8 | 0 |
| 호흡곤란 | 11 | 1 | 8 | 1 |
| 위장관 장애 |
||||
| 오심 | 12 | 0 | 13 | 2 |
| 설사 | 10 | 1 | 13 | 1 |
임상적으로 관련있는 실험실 이상은 표 19에 제시되어 있습니다.
| 실험실 검사 |
Jakafi (N = 165) |
최상의 이용 가능한 치료 (N = 158) |
||
|
모든 등급† |
3등급 이상 |
모든 등급 |
3등급 이상 |
|
| 혈액학 |
||||
| 빈혈 | 82 | 13 | 75 | 8 |
| 호중구 감소증 | 27 | 12 | 23 | 9 |
| 혈소판 감소증 | 58 | 20 | 54 | 17 |
| 화학 |
||||
| 고콜레스테롤혈증 | 88 | 10 | 85 | 8 |
| AST 상승 | 65 | 5 | 54 | 6 |
| ALT 상승 | 73 | 11 | 71 | 16 |
| 감마 글루타밀 트랜스퍼라제 증가 | 81 | 42 | 75 | 38 |
| 크레아티닌 증가 | 47 | 1 | 40 | 2 |
| 리파제 상승 | 38 | 12 | 30 | 9 |
| 아밀라제 상승 | 35 | 8 | 25 | 4 |
6.2 시판 후 경험
다음의 유해 반응은 Jakafi의 시판 후 사용 중에 확인되었습니다. 이러한 반응은 규모가 불확실한 모집단으로부터 자발적으로 보고되었기 때문에, 항상 그 빈도를 신뢰할 수 있게 추정하거나 약물 노출과의 인과 관계를 확립할 수 있는 것은 아닙니다.
- 감염 및 기생충 감염: 단순포진 바이러스 재활성화 및/또는 전파
7. 약물 상호 작용
7.1 Jakafi에 대한 다른 약물의 영향
Fluconazole
Jakafi와 fluconazole의 병용은 ruxolitinib 노출을 증가시켜 [임상 약리학 (12.3) 참조] 노출 관련 부작용의 위험을 증가시킬 수 있습니다. Jakafi와 1일 200mg을 초과하는 fluconazole 용량의 병용을 피하십시오. 200mg 이하의 fluconazole 용량과 병용 투여하는 경우 Jakafi 용량을 줄이십시오 [용법 및 용량 (2.6) 참조].
강력한 CYP3A4 억제제
Jakafi와 강력한 CYP3A4 억제제의 병용은 ruxolitinib 노출을 증가시켜 [임상 약리학 (12.3) 참조] 노출 관련 부작용의 위험을 증가시킬 수 있습니다. aGVHD 또는 cGVHD 환자를 제외하고 강력한 CYP3A4 억제제와 병용 투여하는 경우 Jakafi 용량을 줄이십시오 [용법 및 용량 (2.6) 참조].
강력한 CYP3A4 유도제
Jakafi와 강력한 CYP3A4 유도제의 병용은 ruxolitinib 노출을 감소시켜 [임상 약리학 (12.3) 참조] Jakafi의 효능을 감소시킬 수 있습니다. 환자를 자주 모니터링하고 안전성과 효능에 따라 Jakafi 용량을 조정하십시오 [임상 약리학 (12.3) 참조].
8. 특정 환자군에서의 사용
8.1 임신
위험 요약
임신한 랫트와 토끼에게 기관 형성 기간 동안 ruxolitinib을 투여했을 때 모체 독성과 관련된 용량에서 부작용 발생 결과가 나타났습니다 (데이터 참조). 약물 관련 위험을 알리기 위해 임산부에게 Jakafi를 사용한 연구는 없습니다.
적응증 모집단의 주요 선천적 결손 및 유산에 대한 배경 위험은 알려져 있지 않습니다. 임신 중 부작용 결과는 산모의 건강이나 약물 사용과 관계없이 발생합니다. 미국 일반 인구의 주요 선천적 결손에 대한 배경 위험은 2%~4%이며, 유산은 임상적으로 인지된 임신의 15%~20%입니다.
데이터
동물 데이터
Ruxolitinib은 기관 형성 기간 동안 임신한 랫트 또는 토끼에게 랫트의 경우 15, 30 또는 60mg/kg/일, 토끼의 경우 10, 30 또는 60mg/kg/일 용량으로 경구 투여되었습니다. 치료 관련 기형은 없었습니다. 태아 체중의 약 9% 감소와 같은 부작용 발생 결과는 60mg/kg/일의 최고 용량 및 모체 독성 용량에서 랫트에서 나타났습니다. 이 용량은 최대 권장 용량인 25mg 1일 2회에서의 임상 노출의 약 2배인 노출(AUC)을 초래합니다. 토끼의 경우, 태아 체중의 약 8% 감소 및 후기 재흡수 증가는 60mg/kg/일의 최고 용량 및 모체 독성 용량에서 나타났습니다. 이 용량은 최대 권장 용량에서의 임상 노출의 약 7%입니다.
랫트의 출산 전후 발달 연구에서, 임신한 동물에게 최대 30mg/kg/일 용량의 ruxolitinib을 착상부터 수유까지 투여했습니다. 평가된 최고 용량(최대 권장 용량인 25mg 1일 2회에서의 임상 노출의 34%)에서 새끼의 생식력 지수 또는 모체 또는 배아 태아 생존, 성장 및 발달 매개변수에 대한 약물 관련 부작용 소견은 없었습니다.
8.2 수유
위험 요약
모유에서 ruxolitinib의 존재, 모유 수유아에 대한 영향 또는 모유 생산에 대한 영향에 대한 데이터는 없습니다. Ruxolitinib 및/또는 그 대사체는 수유 중인 랫트의 모유에 존재했습니다(데이터 참조). 많은 약물이 모유에 존재하고 인체 연구에서 Jakafi에 대해 혈소판 감소증 및 빈혈이 나타날 가능성이 있으므로 Jakafi 치료 중 및 최종 용량 투여 후 2주 동안 모유 수유를 중단하십시오.
데이터
동물 데이터
수유 중인 랫트에게 출산 후 10일에 [14C]로 표지된 ruxolitinib(30mg/kg) 단일 용량을 투여한 후 최대 24시간 동안 혈장 및 모유 샘플을 채취했습니다. 모유에서 총 방사능의 AUC는 모체 혈장 AUC의 약 13배였습니다. 추가 분석 결과 모유에서 ruxolitinib 및 여러 대사체가 모두 모체 혈장보다 높은 수준으로 존재하는 것으로 나타났습니다.
8.4 소아 사용
골수섬유증
소아 환자의 골수섬유증 치료를 위한 Jakafi의 안전성과 유효성은 확립되지 않았습니다.
진성 적혈구증가증
소아 환자의 진성 적혈구증가증 치료를 위한 Jakafi의 안전성과 유효성은 확립되지 않았습니다.
급성 이식편대숙주병
스테로이드 불응성 aGVHD 치료를 위한 Jakafi의 안전성과 유효성은 12세 이상의 소아 환자 치료에 대해 확립되었습니다. 스테로이드 불응성 aGVHD 소아 환자에서 Jakafi의 사용은 성인의 적절하고 잘 통제된 Jakafi 시험의 증거 [임상 연구 참조 (14.3)] 및 소아 환자의 추가 약동학 및 안전성 데이터에 의해 뒷받침됩니다. 스테로이드 불응성 aGVHD 치료를 위한 Jakafi의 안전성과 유효성은 12세 미만의 소아 환자에서 확립되지 않았습니다.
만성 이식편대숙주병
1~2차례의 전신 요법 실패 후 cGVHD 치료를 위한 Jakafi의 안전성과 유효성은 12세 이상의 소아 환자 치료에 대해 확립되었습니다. 1~2차례의 전신 요법 실패 후 cGVHD 소아 환자에서 Jakafi의 사용은 성인 및 청소년의 적절하고 잘 통제된 Jakafi 시험의 증거 [임상 연구 참조 (14.4)] 및 소아 환자의 추가 약동학 및 안전성 데이터에 의해 뒷받침됩니다. cGVHD 치료를 위한 Jakafi의 안전성과 유효성은 12세 미만의 소아 환자에서 확립되지 않았습니다.
기타 골수증식성 종양, 백혈병 및 고형 종양
재발성 또는 불응성 고형 종양, 백혈병 또는 골수증식성 종양 환자를 대상으로 한 단일군 시험(NCT01164163)에서 ruxolitinib의 안전성과 유효성을 평가했지만 확립되지는 않았습니다. 환자에는 18명의 어린이(2세~12세 미만)와 14명의 청소년(12세~17세 미만)이 포함되었습니다. 전체적으로 환자의 19%가 1회 이상의 주기를 받았습니다. 이 시험에서 소아 환자에게 새로운 안전성 신호는 관찰되지 않았습니다.
고위험, de novo CRLF2 재배열 또는 JAK 경로 돌연변이 Ph 유사 급성 림프구성 백혈병(ALL) 치료를 위한 ruxolitinib과 화학 요법의 병용 요법의 안전성과 유효성을 단일군 시험(NCT02723994)에서 평가했지만 확립되지는 않았습니다. 환자에는 2명의 유아(2세 미만), 42명의 어린이(2세~12세 미만) 및 62명의 청소년(12세~17세 미만)이 포함되었습니다. 이 시험에서 소아 환자에게 새로운 안전성 신호는 관찰되지 않았습니다.
어린 동물 독성 데이터
ruxolitinib을 어린 쥐에게 투여한 결과 성장 및 뼈 측정에 영향을 미쳤습니다. 생후 7일(인간 신생아에 해당)부터 1.5~75mg/kg/day 용량으로 투여했을 때, ≥ 30mg/kg/day 용량에서 골절 증거가 나타났으며, ≥ 5mg/kg/day 용량에서 체중 및 기타 뼈 측정값[예: 골밀도, 말초 정량 컴퓨터 단층 촬영 및 X선 분석]에 영향을 미쳤습니다. 생후 21일(인간 2~3세에 해당)부터 5~60mg/kg/day 용량으로 투여했을 때, ≥ 15mg/kg/day 용량에서 체중 및 뼈에 영향을 미쳤으며, 60mg/kg/day에서 유해한 것으로 간주되었습니다. 모든 연령대에서 수컷이 암컷보다 더 심각한 영향을 받았으며, 생후 초기 투여를 시작했을 때 일반적으로 영향이 더 심각했습니다. 이러한 결과는 최대 권장 용량인 25mg 1일 2회의 임상 노출의 최소 27%인 노출에서 관찰되었습니다.
8.5 노인 환자에서의 사용
Jakafi 임상 연구의 MF 환자 총 수 중 52%는 65세 이상이었고 15%는 75세 이상이었습니다. 이러한 환자와 젊은 환자 사이에 Jakafi의 안전성 또는 유효성에 전반적인 차이는 관찰되지 않았습니다.
aGVHD 환자를 대상으로 한 Jakafi 임상 연구에는 65세 이상의 피험자가 충분하지 않아 젊은 피험자와 다르게 반응하는지 여부를 판단할 수 없었습니다.
임상 시험에서 Jakafi로 치료받은 cGVHD 환자 총 수 중 11%는 65세 이상이었습니다. 이러한 환자와 젊은 환자 사이에 Jakafi의 안전성 또는 유효성에 전반적인 차이는 관찰되지 않았습니다.
8.6 신장애
ruxolitinib 및 활성 대사체의 총 노출은 중등도(CLcr 30~59mL/min) 및 중증(CLcr 15~29mL/min) 신장애, 그리고 투석을 받는 ESRD(CLcr 15mL/min 미만) 환자에서 증가했습니다 [see Clinical Pharmacology (12.3)]. 권장대로 Jakafi 용량을 조정하십시오 [see Dosage and Administration (2.7)].
8.7 간장애
ruxolitinib의 노출은 경증(Child-Pugh A), 중등도(Child-Pugh B) 및 중증(Child-Pugh C) 간장애 환자에서 증가했습니다 [see Clinical Pharmacology (12.3)].
간장애가 있는 MF 또는 PV 환자의 경우 권장대로 Jakafi 용량을 줄이십시오 [see Dosage and Administration (2.7)]. 4기 간 aGVHD 환자의 경우 권장대로 Jakafi 용량을 줄이십시오.
Score 3 간 cGVHD 환자의 경우 독성에 대해 더 자주 혈구 수를 모니터링하고 부작용이 발생하면 Jakafi 용량을 조정하십시오 [see Dosage and Administration (2.7) and Clinical Pharmacology (12.3)].
10. 과량 복용
Jakafi 과다복용에 대한 특별한 해독제는 없습니다. 최대 200mg의 단회 용량은 허용 가능한 급성 내약성을 보였습니다. 권장 용량보다 높은 반복 용량은 백혈구 감소증, 빈혈 및 혈소판 감소증을 포함한 골수억제 증가와 관련이 있습니다. 적절한 지지 치료를 해야 합니다.
혈액 투석은 Jakafi의 배설을 향상시킬 것으로 예상되지 않습니다.
11. 제품 설명
Ruxolitinib phosphate는 (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate라는 화학명을 가진 키나아제 억제제이며, 분자량은 404.36입니다. Ruxolitinib phosphate의 구조식은 다음과 같습니다:
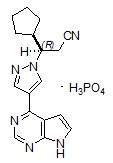
Ruxolitinib phosphate는 흰색에서 회백색 또는 연분홍색 분말이며 pH 1~8의 수성 완충액에 용해됩니다.
Jakafi (ruxolitinib) 정제는 경구 투여용입니다. 각 정제는 6.6mg, 13.2mg, 19.8mg, 26.4mg 또는 33mg의 ruxolitinib phosphate를 함유하고 있으며, 이는 각각 5mg, 10mg, 15mg, 20mg 또는 25mg의 ruxolitinib 유리 염기에 해당합니다. 또한 미결정 셀룰로오스, 락토오스 일수화물, 스테아르산마그네슘, 콜로이드성 이산화규소, 글리콜산 나트륨 전분, 포비돈 및 히드록시프로필 셀룰로오스를 함유합니다.
12. 임상 약리학
12.1 작용 기전
키나아제 억제제인 Ruxolitinib은 조혈 및 면역 기능에 중요한 여러 사이토카인과 성장 인자의 신호 전달을 매개하는 Janus Associated Kinases (JAKs) JAK1 및 JAK2를 억제합니다. JAK 신호 전달은 사이토카인 수용체에 대한 STAT(신호 변환기 및 전사 활성화제)의 모집, 활성화 및 후속적인 STAT의 핵으로의 국소화를 포함하며, 이는 유전자 발현의 조절로 이어집니다.
MF 및 PV는 JAK1 및 JAK2 신호 전달 장애와 관련된 것으로 알려진 골수증식성 종양(MPN)입니다. JAK2V617F 양성 MPN 마우스 모델에서 ruxolitinib의 경구 투여는 비장 비대를 예방하고, 비장에서 JAK2V617F 돌연변이 세포를 우선적으로 감소시키고, 순환하는 염증성 사이토카인(예: TNF-α, IL-6)을 감소시켰습니다.
JAK-STAT 신호 전달 경로는 GVHD 발병 기전에 중요한 여러 면역 세포 유형의 발달, 증식 및 활성화를 조절하는 역할을 합니다. aGVHD 마우스 모델에서 ruxolitinib의 경구 투여는 결장 균질액에서 염증성 사이토카인의 발현 감소 및 결장에서 면역 세포 침윤 감소와 관련이 있었습니다.
12.2 약력학
Jakafi는 MF 및 PV 환자의 전혈에서 사이토카인 유도 STAT3 인산화를 억제합니다. STAT3 인산화는 Jakafi 투여 후 2시간 후에 최대 억제에 도달했으며 MF 및 PV 환자에서 10시간까지 거의 기준치로 돌아왔습니다.
심장 전기생리학
최고 권장 시작 용량의 1.25~10배 용량에서 Jakafi는 임상적으로 관련 있는 정도까지 QT 간격을 연장하지 않습니다.
12.3 약동학
평균 ruxolitinib 최대 혈장 농도(Cmax) 및 AUC는 5 mg~200 mg(승인된 최고 권장 총 1일 용량 25mg 1일 2회의 4배)의 단일 용량 범위에서 비례적으로 증가했습니다. 평균 ruxolitinib Cmax는 5 mg~200 mg의 단일 용량 범위에서 205 nM~7100 nM였으며 AUC는 862 nM*hr~30700 nM*hr였습니다.
흡수
Ruxolitinib은 투여 후 1~2시간 이내에 Cmax에 도달합니다. Ruxolitinib의 경구 흡수율은 최소 95%로 추정됩니다.
음식의 영향
고지방, 고칼로리 식사(약 800~1000칼로리, 그 중 50%는 지방에서 유래)와 함께 Jakafi를 투여했을 때 ruxolitinib의 약동학에서 임상적으로 관련 있는 변화는 관찰되지 않았습니다.
분포
MF 환자에서 평균 ruxolitinib 정상 상태 분포 용적은 72L(변동 계수[CV] 29%)이고 PV 환자에서는 75L(23%)입니다.
Ruxolitinib의 단백질 결합은 약 97%이며, 대부분 알부민에 결합합니다.
제거
건강한 지원자에서 ruxolitinib의 평균 제거 반감기는 약 3시간이며 ruxolitinib 및 대사체의 평균 제거 반감기는 약 5.8시간입니다.
Ruxolitinib 클리어런스(%CV)는 MF 여성에서 17.7L/h, 남성에서 22.1L/h(39%)였습니다.
Ruxolitinib 클리어런스(%CV)는 PV 환자에서 12.7L/h(42%)였습니다.
Ruxolitinib 클리어런스(%CV)는 aGVHD 환자에서 11.8L/h(63%)였습니다.
Ruxolitinib 클리어런스(%CV)는 cGVHD 환자에서 9.7L/h(51%)였습니다.
대사
Ruxolitinib은 CYP3A4에 의해 대사되며, CYP2C9에 의해서는 덜 대사됩니다.
배설
방사성 표시된 ruxolitinib을 단일 경구 투여한 후 방사능의 74%는 소변으로, 22%는 대변으로 배설되었습니다. 변하지 않은 약물은 배설된 총 방사능의 1% 미만을 차지했습니다.
특정 집단
연령(12~73세), 인종(백인, 아시아인), 성별 또는 체중(29~139kg)에 따라 ruxolitinib 약동학에서 임상적으로 관련 있는 차이는 관찰되지 않았습니다.
신장애 환자
정상 신기능(CLcr ≥ 90mL/min)을 가진 환자에 비해 경증, 중등도, 중증 신장애 및 투석 후 ESRD 환자에서 ruxolitinib 및 활성 대사체의 총 AUC는 각각 1.3배, 1.5배, 1.9배 및 1.6배 증가했습니다. 약력학적 마커인 pSTAT3 억제의 변화는 신장애로 인한 대사체 노출 증가와 일치했습니다. Ruxolitinib은 투석으로 제거되지 않습니다. 그러나 투석에 의한 일부 활성 대사체의 제거 가능성은 배제할 수 없습니다.
간장애 환자
aGVHD 또는 cGVHD 환자에서 NCI 기준(총 빌리루빈 > ULN 및 AST)에 따른 경증~중증 간장애에 따라 ruxolitinib 약동학에 임상적으로 관련 있는 영향은 관찰되지 않았습니다.
정상 간 기능을 가진 환자에 비해 경증(Child-Pugh A), 중등도(Child-Pugh B) 및 중증(Child-Pugh C) 간장애 환자에서 ruxolitinib AUC는 각각 1.9배, 1.3배 및 1.7배 증가했습니다.
약력학적 마커인 pSTAT3 억제의 변화는 ruxolitinib 노출 증가와 일치했지만, 중증 간장애 코호트에서는 일부 환자에서 약력학적 활성이 ruxolitinib의 혈장 농도에 따라 예상보다 더 오래 지속되었습니다.
이식편대숙주병의 간 관련 환자
1, 2 또는 3기 간 aGVHD 또는 1 또는 2점 간 cGVHD에 따라 ruxolitinib 약동학에 임상적으로 관련 있는 영향은 관찰되지 않았습니다.
간 aGVHD가 없는 환자에 비해 4기 간 aGVHD 환자에서 ruxolitinib의 겉보기 클리어런스가 더 낮게 관찰되었습니다.
3점 간 cGVHD가 ruxolitinib의 약동학에 미치는 영향은 알려져 있지 않습니다.
약물 상호작용 연구
임상 연구 및 모델 기반 접근법
Fluconazole: Fluconazole 100 ~ 400mg을 1일 1회 투여 시(중등도 CYP3A4 및 CYP2C9 억제제) 정상 상태 ruxolitinib AUC가 약 100% ~ 300% 증가합니다. [용법 및 용량 (2.6) 및 약물 상호작용 (7) 참조].
강력 CYP3A4 억제제: Ketoconazole(강력 CYP3A4 억제제)은 ruxolitinib Cmax 를 33%, AUC를 91% 증가시켰으며, ruxolitinib 반감기를 3.7시간에서 6시간으로 연장시켰습니다. [용법 및 용량 (2.6) 및 약물 상호작용 (7) 참조].
중등도 CYP3A4 억제제: Erythromycin(중등도 CYP3A4 억제제)은 ruxolitinib Cmax 를 8%, AUC를 27% 증가시켰습니다. [약물 상호작용 (7) 참조].
강력 CYP3A4 유도제: Rifampin(강력 CYP3A4 유도제)은 ruxolitinib Cmax 를 32%, AUC를 61% 감소시켰습니다. ruxolitinib의 활성 대사체에 대한 상대적 노출은 약 100% 증가했습니다. [약물 상호작용 (7) 참조].
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Ruxolitinib 및 그 M18 대사체는 CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 또는 CYP3A4를 억제하지 않았습니다. Ruxolitinib은 임상적으로 관련 있는 농도에서 CYP1A2, CYP2B6 또는 CYP3A4를 유도하지 않았습니다.
Transporter Systems: Ruxolitinib 및 그 M18 대사체는 임상적으로 관련 있는 농도에서 P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1 또는 OAT3을 억제하지 않았습니다. Ruxolitinib은 P-gp 기질이 아니었습니다.
13. 비임상 독성학
13.1 발암성, 돌연변이원성, 생식능력 저하
룩솔리티닙은 6개월 Tg.rasH2 형질전환 마우스 모델 또는 2년간의 랫트 발암성 연구에서 발암성이 없었습니다.
룩솔리티닙은 박테리아 돌연변이원성 분석(Ames test)에서 돌연변이원성이 없었으며, in vitro 염색체 이상 분석(배양된 사람 말초혈액 림프구) 또는 in vivo 랫트 골수 미세핵 분석에서 클라스토젠성이 없었습니다.
생식능력 연구에서, 룩솔리티닙은 교배 전과 교배 기간 동안 수컷 랫트에, 그리고 교배 전부터 착상일(임신 7일째)까지 암컷 랫트에 투여되었습니다. 룩솔리티닙은 10, 30 또는 60 mg/kg/일 용량에서 수컷 또는 암컷 랫트의 생식능력 또는 생식 기능에 영향을 미치지 않았습니다. 그러나, 암컷 랫트에서 30 mg/kg/일 이상의 용량은 착상 후 손실을 증가시켰습니다. 30 mg/kg/일 용량에서의 노출(AUC)은 1일 2회 최대 권장 용량 25 mg의 임상 노출량의 약 34%입니다.
14. 임상 연구
14.1 골수섬유증
골수섬유증(원발성 골수섬유증, 진성적혈구증가증 후 골수섬유증 또는 본태성혈소판증가증 후 골수섬유증) 환자를 대상으로 두 건의 무작위 배정 3상 연구(연구 1 및 연구 2)가 수행되었습니다. 두 연구 모두에서 환자는 촉진 가능한 비장비대가 갈비뼈 아래쪽 가장자리에서 최소 5cm 이상이었고, 국제 실무 그룹 합의 기준(IWG)에 따라 위험 범주가 중등도 2(예후 인자 2개) 또는 고위험(예후 인자 3개 이상)이었습니다.
Jakafi의 시작 용량은 혈소판 수치를 기준으로 했습니다. 혈소판 수치가 100~200 x 109/L인 환자는 Jakafi 15 mg을 1일 2회 복용하기 시작했고, 혈소판 수치가 200 x 109/L 초과인 환자는 Jakafi 20 mg을 1일 2회 복용하기 시작했습니다. 그런 다음 용량은 내약성과 효능에 따라 개별적으로 조정되었으며, 혈소판 수치가 100~125 x 109/L인 환자의 최대 용량은 1일 2회 20 mg, 혈소판 수치가 75~100 x 109/L인 환자의 최대 용량은 1일 2회 10 mg, 혈소판 수치가 50~75 x 109/L인 환자의 최대 용량은 1일 2회 5 mg이었습니다.
연구 1
연구 1(NCT00952289)은 기존 치료법에 불응하거나 적합하지 않은 309명의 환자를 대상으로 한 이중맹검, 무작위 배정, 위약 대조 연구였습니다. 평균 연령은 68세(범위 40~91세)였으며, 환자의 61%가 65세 이상이었고 54%는 남성이었습니다. 환자의 50%는 원발성 골수섬유증, 31%는 진성적혈구증가증 후 골수섬유증, 18%는 본태성혈소판증가증 후 골수섬유증이었습니다. 환자의 21%는 연구 등록 후 8주 이내에 적혈구 수혈을 받았습니다. 평균 헤모글로빈 수치는 10.5g/dL였고 평균 혈소판 수치는 251 x 109/L였습니다. 환자의 평균 촉진 가능한 비장 길이는 갈비뼈 아래쪽 가장자리에서 16cm였으며, 81%는 비장 길이가 갈비뼈 아래쪽 가장자리에서 10cm 이상이었습니다. 자기공명영상(MRI) 또는 컴퓨터단층촬영(CT)으로 측정한 환자의 평균 비장 부피는 2595cm3(범위 478cm3~8881cm3)였습니다. (정상 상한선은 약 300cm3입니다.)
환자들은 Jakafi 또는 대응 위약을 투여받았습니다. 주요 효능 평가 변수는 MRI 또는 CT로 측정했을 때 24주차에 비장 부피가 기준치 대비 35% 이상 감소한 환자의 비율이었습니다.
이차 평가 변수에는 비장 부피가 35% 이상 감소한 기간과 수정된 골수섬유증 증상 평가 양식(MFSAF) v2.0 일지에 따라 측정했을 때 기준치 대비 24주차까지 총 증상 점수가 50% 이상 감소한 환자의 비율이 포함되었습니다.
연구 2
연구 2(NCT00934544)는 219명의 환자를 대상으로 한 공개, 무작위 배정 연구였습니다. 환자들은 Jakafi 대 최적의 기존 치료법에 2:1로 무작위 배정되었습니다. 최적의 기존 치료법은 연구자가 환자별로 선택했습니다. 최적의 기존 치료법군에서 환자의 10% 이상이 투여받은 약물은 하이드록시유레아(47%)와 글루코코르티코이드(16%)였습니다. 평균 연령은 66세(범위 35~85세)였으며, 환자의 52%가 65세 이상이었고 57%는 남성이었습니다. 환자의 53%는 원발성 골수섬유증, 31%는 진성적혈구증가증 후 골수섬유증, 16%는 본태성혈소판증가증 후 골수섬유증이었습니다. 환자의 21%는 연구 등록 후 8주 이내에 적혈구 수혈을 받았습니다. 평균 헤모글로빈 수치는 10.4g/dL였고 평균 혈소판 수치는 236 x 109/L였습니다. 환자의 평균 촉진 가능한 비장 길이는 갈비뼈 아래쪽 가장자리에서 15cm였으며, 70%는 비장 길이가 갈비뼈 아래쪽 가장자리에서 10cm 이상이었습니다. MRI 또는 CT로 측정한 환자의 평균 비장 부피는 2381cm3(범위 451cm3~7765cm3)였습니다.
주요 효능 평가 변수는 MRI 또는 CT로 측정했을 때 48주차에 비장 부피가 기준치 대비 35% 이상 감소한 환자의 비율이었습니다.
연구 2의 이차 평가 변수는 MRI 또는 CT로 측정했을 때 기준치 대비 24주차까지 비장 부피가 35% 이상 감소한 환자의 비율이었습니다.
연구 1 및 2 효능 결과
연구 1 및 2의 주요 평가 변수에 대한 효능 분석은 아래 표 20에 제시되어 있습니다. 연구 1의 위약군 및 연구 2의 최적의 기존 치료법군과 비교했을 때 두 연구 모두에서 Jakafi군에서 유의하게 더 많은 비율의 환자에서 기준치 대비 비장 부피가 35% 이상 감소했습니다. Jakafi군에서 유사한 비율의 환자에서 촉진 가능한 비장 길이가 50% 이상 감소했습니다.
| 연구 1 | 연구 2 | |||
| Jakafi (N=155) |
위약 (N=154) |
Jakafi (N=146) |
최적의 기존 치료법 (N=73) |
|
| 시점 | 24주차 | 48주차 | ||
| 비장 부피 35% 이상 감소 환자 수 (%) | 65 (42) | 1 (< 1) | 41 (29) | 0 |
| P-value | < 0.0001 | < 0.0001 | ||
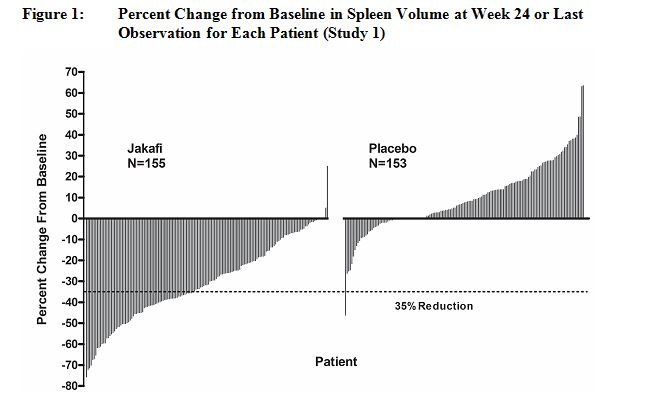
그림 1은 24주차(Jakafi N=139, placebo N=106) 또는 무작위 배정 치료 24주를 완료하지 않은 환자의 경우 24주차 이전의 마지막 평가 시점(Jakafi N=16, placebo N=47)에서 각 환자의 비장 부피 기준치 대비 백분율 변화를 보여줍니다. 기준 비장 부피가 누락된 환자 1명(placebo)은 포함되지 않았습니다.
연구 1에서 MF 증상은 2차 평가변수였으며 수정된 골수섬유증 증상 평가 양식(MFSAF) v2.0 일지을 사용하여 측정되었습니다. 수정된 MFSAF는 MF의 핵심 증상(복부 불편, 왼쪽 갈비뼈 아래 통증, 야간 발한, 가려움증, 뼈/근육 통증 및 조기 포만감)을 기록하는 일일 일지입니다. 증상 점수는 0에서 10까지의 범위였으며, 0은 증상 “없음”을 나타내고 10은 “상상할 수 있는 최악의” 증상을 나타냅니다. 이 점수들을 합산하여 최대 60점인 일일 총점을 산출했습니다.
표 21은 연구 1에서 기준치부터 24주차까지의 총 증상 점수 평가를 제시하며, 여기에는 최소 50% 감소(즉, 증상 개선)를 보인 환자의 비율이 포함됩니다. 기준 시점에서 평균 총 증상 점수는 Jakafi 그룹에서 18.0, placebo 그룹에서 16.5였습니다. Jakafi 그룹에서 총 증상 점수가 50% 이상 감소한 환자의 비율이 placebo 그룹보다 높았으며, 반응까지의 중앙값 시간은 4주 미만이었습니다.
| Jakafi (N=148) |
Placebo (N=152) |
|
| 24주차까지 총 증상 점수가 50% 이상 감소한 환자 수 (%) | 68 (46) | 8 (5) |
| P-value | < 0.0001 | |
그림 2는 24주차(자카피 N=129, 위약 N=103) 또는 24주 무작위 치료를 완료하지 않은 환자의 경우 24주 이전 무작위 치료의 마지막 평가 시점(자카피 N=16, 위약 N=42)에 각 환자의 총 증상 점수 기준 변화율을 보여줍니다. 기준 총 증상 점수가 0인 환자 5명, 기준 데이터가 없는 환자 8명, 기준 이후 데이터가 불충분한 환자 6명은 결과에서 제외되었습니다.
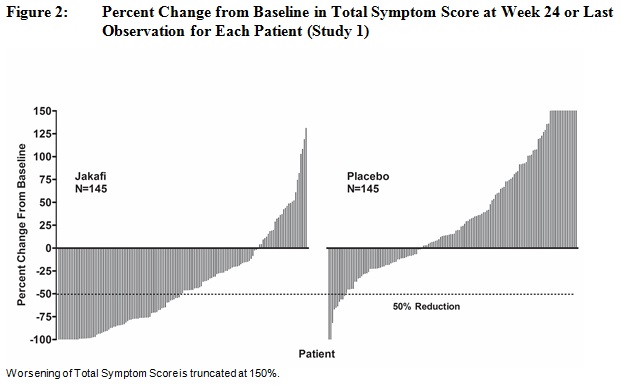
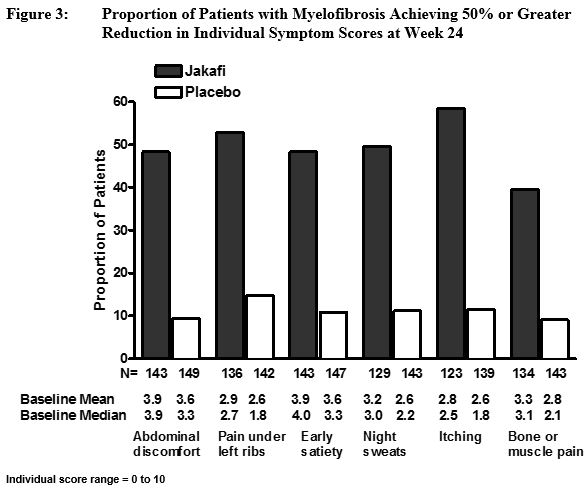
그림 3은 총 증상 점수를 구성하는 개별 증상 각각에서 50% 이상 개선된 환자의 비율을 보여주며, 자카피 치료군에서 총 증상 점수 반응률이 더 높은 데에는 6가지 증상 모두가 기여했음을 나타냅니다.
자카피를 투여받은 환자에 대한 탐색적 분석에서 PROMIS® Fatigue 7-item short form 총점으로 측정한 24주차의 피로 관련 증상(예: 피로, 탈진, 정신적 피로, 에너지 부족) 및 일상 활동에 대한 관련 영향(예: 직장, 자기 관리, 운동과 관련된 활동 제한)의 개선도 나타났습니다. 기준에서 24주차까지 PROMIS® Fatigue 총점이 4.5점 이상 감소한 환자는 피로 반응을 달성한 것으로 간주되었습니다. 피로 반응은 자카피군 환자의 35%에서 보고되었으며, 위약군 환자의 14%에서 보고되었습니다.
전체 생존율은 연구 1과 연구 2 모두에서 2차 평가변수였습니다. 대조군의 환자는 두 연구 모두에서 교차 연구에 참여할 수 있었으며, 교차 연구까지의 중앙값 시간은 연구 1에서 9개월, 연구 2에서 17개월이었습니다.
그림 4와 그림 5는 연구에 남아 있는 모든 환자가 144주 동안 연구를 완료한 후 전향적으로 계획된 분석에서 전체 생존율의 Kaplan-Meier 곡선을 보여줍니다.
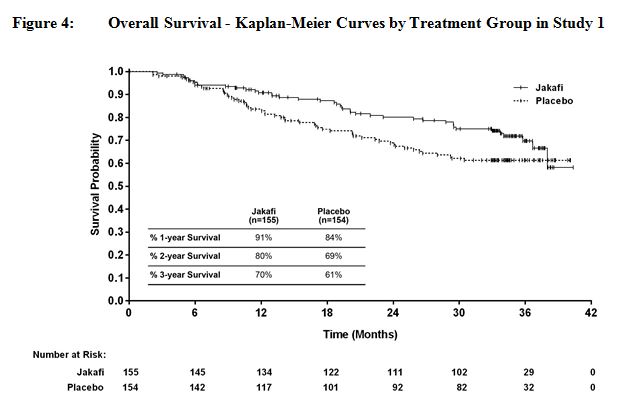
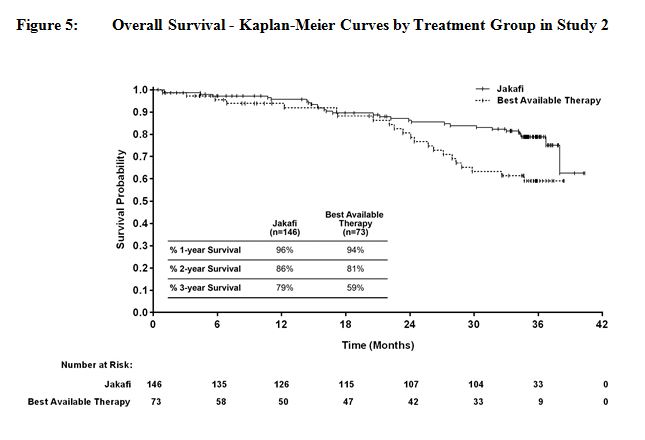
14.2 진성적혈구증가증
연구 3(NCT01243944)은 PV 환자 222명을 대상으로 실시된 무작위, 공개, 활성 대조군 제3상 연구였습니다. 환자들은 최소 24주 동안 PV 진단을 받았고, 하이드록시우레아에 대한 반응이 불충분하거나 내약성이 없었으며, 사혈이 필요했고, 비장비대증을 보였습니다. 모든 환자는 무작위 배정 전 40~45%의 헤마토크릿 조절을 입증해야 했습니다. 연령은 33세에서 90세였으며, 환자의 30%가 65세 이상이었고 66%가 남성이었습니다. 환자의 MRI 또는 CT로 측정한 중앙값 비장 용적은 1272cm3(범위 254cm3~5147cm3)였고, 늑골연 아래에서 촉진되는 중앙값 비장 길이는 7cm였습니다.
환자들은 자카피 또는 최적의 치료법으로 무작위 배정되었습니다. 자카피의 시작 용량은 1일 2회 10mg이었습니다. 그런 다음 용량은 내약성과 효능에 따라 개별화되었으며 최대 용량은 1일 2회 25mg이었습니다. 32주차에 98명의 환자가 여전히 자카피를 복용하고 있었으며, 8%는 1일 2회 20mg 이상, 15%는 1일 2회 20mg, 33%는 1일 2회 15mg, 34%는 1일 2회 10mg, 10%는 1일 2회 10mg 미만을 복용했습니다. 최적의 치료법(BAT)은 연구자가 환자별로 선택했으며 하이드록시우레아(60%), 인터페론/페길화 인터페론(12%), 아나그렐리드(7%), 피포브로만(2%), 레날리도마이드/탈리도마이드(5%), 관찰(15%)이 포함되었습니다.
1차 평가변수는 32주차에 반응을 달성한 환자의 비율이었으며, 반응은 헤마토크릿 조절(8주차 방문부터 시작하여 32주차까지 사혈 자격 없음)과 비장 용적 감소(32주차에 기준 대비 비장 용적 35% 이상 감소)를 모두 달성한 것으로 정의되었습니다. 사혈 자격은 기준에서 얻은 헤마토크릿보다 최소 3% 이상 높은 45% 이상의 확인된 헤마토크릿 또는 48% 이상의 확인된 헤마토크릿 중 낮은 쪽으로 정의되었습니다. 2차 평가변수에는 무작위 배정된 모든 환자 중 1차 평가변수를 달성하고 무작위 배정 후 48주 동안 반응을 유지한 환자의 비율, 그리고 32주차에 완전한 혈액학적 관해를 달성한 환자의 비율이 포함되었으며, 완전한 혈액학적 관해는 헤마토크릿 조절, 혈소판 수 400 x 109/L 이하, 백혈구 수 10 x 109/L 이하를 달성한 것으로 정의되었습니다.
1차 및 2차 평가변수의 결과는 표 22에 제시되어 있습니다. 자카피군의 유의하게 더 많은 비율의 환자가 최적의 치료법에 비해 32주차에 1차 평가변수에 대한 반응을 달성했으며 무작위 배정 후 48주 동안 반응을 유지했습니다. 최적의 치료법에 비해 자카피군의 유의하게 더 많은 비율의 환자도 32주차에 완전한 혈액학적 관해를 달성했습니다.
| 1차 반응은 8주차 방문부터 시작하여 32주차까지 사혈 자격 없음과 32주차에 기준 대비 비장 용적 35% 이상 감소를 모두 달성한 것으로 정의됩니다. | ||
| 자카피 (N=110) |
최적의 치료법 (N=112) |
|
| 32주차에 1차 반응을 달성한 환자 수 (%) | 25 (23%) | 1 (< 1%) |
| 95% CI of the response rate (%) | (15%, 32%) | (0%, 5%) |
| P-value | < 0.0001 | |
| 48주차에 지속적인 1차 반응을 달성한 환자 수 (%) | 22 (20%) | 1 (< 1%) |
| 95% CI of the response rate (%) | (13%, 29%) | (0%, 5%) |
| P-value | < 0.0001 | |
| 32주차에 완전한 혈액학적 관해를 달성한 환자 수 (%) | 26 (24%) | 9 (8%) |
| 95% CI of the response rate (%) | (16%, 33%) | (4%, 15%) |
| P-value | 0.0016 | |
반응의 지속성을 평가하기 위한 연구 3에 대한 추가 분석은 80주차에 Jakafi군에서만 수행되었습니다. 이 군에서 80주차 데이터 마감 시점에 91명(83%)의 환자가 여전히 치료를 받고 있었습니다. 32주차에 1차 반응을 달성한 25명의 환자 중 19명(반응자의 76%)이 80주차까지 반응을 유지했으며, 32주차에 완전한 혈액학적 관해를 달성한 26명의 환자 중 15명(반응자의 58%)이 80주차까지 반응을 유지했습니다.
1차 평가변수를 구성하는 개별 구성 요소에 대한 평가에서, 32주차에 Jakafi군의 66명(60%) 환자에서 헤마토크릿 조절이 이루어진 반면, 최적의 치료를 받은 환자군에서는 21명(19%)이었습니다. Jakafi군의 51명(헤마토크릿 반응자의 77%) 환자는 80주차까지 헤마토크릿 조절을 유지했습니다. 32주차에 Jakafi군의 44명(40%) 환자에서 기준치 대비 비장 용적 감소가 35% 이상이었던 반면, 최적의 치료를 받은 환자군에서는 1명(< 1%)이었습니다. Jakafi군의 43명(비장 용적 감소 반응자의 98%) 환자는 80주차까지 비장 용적 감소를 유지했습니다.
14.3
급성 이식편대숙주병
연구 4(NCT02953678)는 동종 조혈모세포 이식 후 발생하는 스테로이드 불응성 aGVHD 2~4등급(Mount Sinai Acute GVHD International Consortium (MAGIC) 기준) 환자의 Jakafi 치료에 대한 공개 라벨, 단일군, 다기관 연구였습니다. Jakafi는 1일 2회 5mg씩 투여되었으며, 독성이 없는 경우 3일 후 1일 2회 10mg으로 증량할 수 있었습니다.
스테로이드 단독요법에 불응성인 aGVHD 환자는 49명이었습니다. 이 환자들의 평균 연령은 57세(범위, 18-72세)였고, 47%는 남성이었으며, 92%는 백인이었고, 14%는 히스패닉이었습니다. 기준 시점에서 aGVHD는 2등급이 27%, 3등급이 55%, 4등급이 18%였습니다. 84%는 내장 GVHD가 있었습니다. MAGIC 바이오마커 점수의 중앙값은 0.47(범위, 0.10-0.92)였고, ST2 수치의 중앙값은 334mcg/L(범위, 55-1286mcg/L)였습니다. 기준 시점에서 이전 코르티코스테로이드 노출 기간의 중앙값은 15일(범위: 3-106일)이었습니다.
Jakafi의 효능은 28일차 전체 반응률(ORR)(Center for International Blood and Marrow Transplant Research (CIBMTR) 기준에 따른 완전 반응, 매우 좋은 부분 반응 또는 부분 반응) 및 반응 지속 기간을 기반으로 했습니다. ORR 결과는 표 23에 제시되어 있습니다. 28일차 ORR은 2등급 GVHD의 경우 100%, 3등급 GVHD의 경우 40.7%, 4등급 GVHD의 경우 44.4%였습니다.
28일차 반응에서 진행, aGVHD에 대한 새로운 구제 요법 또는 모든 원인으로 인한 사망까지(진행은 이전 반응 평가와 비교하여 다른 장기의 개선 없이 모든 장기에서 한 단계 악화되는 것으로 정의됨) 계산된 반응 지속 기간의 중앙값은 16일(95% CI 9, 83)이었습니다. 또한 28일차 반응자의 경우 28일차 반응에서 사망 또는 aGVHD에 대한 새로운 치료 필요(추가 구제 요법 또는 스테로이드 증량)까지의 시간의 중앙값은 173일(95% CI 66, NE)이었습니다.
| 스테로이드 단독요법 불응성 (n=49) |
|
| 전체 반응 (%) (95% CI) | 28 (57.1%) (42.2, 71.2) |
| 완전 반응 | 15 (30.6%) |
| 매우 좋은 부분 반응 | 2 (4.1%) |
| 부분 반응 | 11 (22.4%) |
14.4 만성 이식편대숙주병
연구 5(REACH-3; NCT03112603)는 동종 조혈모세포 이식 후 코르티코스테로이드 불응성 cGVHD 치료를 위해 Jakafi를 최적의 치료법(BAT)과 비교한 무작위, 공개형, 다기관 연구였습니다. 적격 환자는 NIH Consensus Criteria에 정의된 중등도 또는 중증 cGVHD가 있는 12세 이상 환자로, 코르티코스테로이드 치료 실패 후 추가 치료가 필요하고 추가적인 구제 치료는 1회 이하였습니다. ANC < 1 Gi/L 및 혈소판 수 < 25 Gi/L, 추정 크레아티닌 청소율 < 30 ml/min, 진행성 발병 cGVHD, 산소 포화도 < 90%, 총 빌리루빈 > 2 mg/dL 또는 GVHD로 인한 설사가 있는 환자는 제외되었습니다.
총 329명의 환자가 Jakafi 10mg 1일 2회 투여(n=165) 또는 BAT(n=164)를 1:1로 무작위 배정받았습니다. BAT는 무작위 배정 전에 연구자가 선택했으며 체외 광화학요법(ECP), 저용량 메토트렉세이트(MTX), 미코페놀레이트 모페틸(MMF), mTOR 억제제(에베롤리무스 또는 시롤리무스), 인플릭시맙, 리툭시맙, 펜토스타틴, 이마티닙 또는 이브루티닙 치료를 포함했습니다. 무작위 배정은 cGVHD 중증도(중등도 대 중증)에 따라 계층화되었습니다. 7주기 1일차부터 BAT에 무작위 배정된 환자는 질병 진행, 혼합 반응, 변화 없는 반응, cGVHD 재발 또는 BAT에 대한 독성이 있는 경우 Jakafi로 교차할 수 있었습니다. 모든 환자는 항감염제를 포함한 표준 지지 요법도 받았습니다. 무작위 배정 전에 시작된 GVHD 예방 및 cGVHD 치료 약물(전신 코르티코스테로이드, 칼시뉴린 억제제, 국소 또는 흡입 코르티코스테로이드 요법 포함)은 기관 지침에 따라 계속 투여할 수 있었습니다. 표 24는 무작위 배정된 모집단의 인구 통계 및 기준 질병 특성을 보여줍니다.
|
||
|
Jakafi |
최적의 치료법 |
|
| 중앙값 연령, 세(범위) | 49 (13, 73) | 50 (12, 76) |
|
12세 ~ < 18세, n (%) |
4 (2) | 8 (5) |
|
65세 초과, n (%) |
18 (11) | 22 (13) |
| 남성, n (%) | 109 (66) | 92 (56) |
| 인종, n (%) | ||
|
백인 |
116 (70) | 132 (81) |
|
흑인 |
2 (1) | 0 |
|
아시아인 |
33 (20) | 21 (13) |
|
아메리카 원주민 또는 알래스카 원주민 |
2 (1) | 0 |
|
기타 |
9 (6) | 4 (2) |
|
알 수 없음 |
3 (2) | 7 (4) |
| cGVHD 진단부터 무작위 배정까지의 중앙값(범위) 시간(일) | 174 (7-2017) | 150 (10-1947) |
| 이전 치료 | ||
|
cGVHD에 대한 이전 치료 없음 |
2 (1) | 1 (1) |
|
1차 스테로이드 단독요법 실패 |
115 (70) | 125 (76) |
|
스테로이드를 포함한 1차 병용요법 실패 |
42 (25) | 30 (18) |
|
2차 요법 실패 |
6 (4) | 8 (5) |
| 4개 이상의 장기 관련, n (%) | 67 (41) | 63 (38) |
| 중증 cGVHD, n (%) | 86 (52) | 79 (48) |
| cGVHD 총 증상 점수 중앙값 (범위) | 19 (0-80) | 18 (1-54) |
| 기준 코르티코스테로이드 용량 중앙값 (범위) (PE mg/kg)* | 0.29 (0.01-1.81) | 0.26 (0.06-1.21) |
Jakafi의 효능은 2014 NIH 반응 기준에 따른 전체 반응률(ORR)(완전 반응 또는 부분 반응 포함) 및 반응 지속 기간을 기반으로 7주기 1일차까지 평가되었습니다. ORR 결과는 표 25에 제시되어 있습니다. Jakafi와 BAT군 간의 ORR 차이는 13%(95% CI 3%, 23%)였습니다. 반응을 보인 환자에서 첫 반응까지의 중앙값 시간은 Jakafi군에서 3주(범위, 2~24주), BAT군에서 4주(범위, 2~25주)였습니다. 첫 반응부터 진행, 사망 또는 cGVHD에 대한 새로운 전신 치료까지 계산된 반응 지속 기간의 중앙값은 Jakafi군에서 4.2개월(95% CI 3.2, 6.7), BAT군에서 2.1개월(95% CI 1.6, 3.2)이었으며, 첫 반응부터 사망 또는 cGVHD에 대한 새로운 전신 치료까지의 중앙값 시간은 Jakafi군에서 25개월(95% CI 16.8, NE), BAT군에서 5.6개월(95% CI 4.1, 7.8)이었습니다.
|
||
| Jakafi (N=165) |
최적의 유효 치료법 (N=164) |
|
| 전체 반응률 (%) (95% CI)* |
116 (70%) (63%, 77%) |
94 (57%) (49%, 65%) |
| 완전 반응 (%) | 14 (8%) | 8 (5%) |
| 부분 반응 (%) | 102 (62%) | 86 (52%) |
ORR 결과는 7주기 1일차까지 cGVHD 총 증상 점수가 7점 이상 감소한 환자에 대한 탐색적 분석으로 뒷받침되었으며, Jakafi군에서는 66명(40%; 95% CI 32, 48), BAT군에서는 47명(29%; 95% CI 22, 36)이었습니다.
16. 공급 형태/보관 및 취급
Jakafi (룩솔리티닙) 정제는 다음과 같이 제공됩니다:
| 실온 20°C~25°C (68°F~77°F)에 보관하십시오. 15°C~30°C (59°F~86°F)의 온도 변화는 허용됩니다 [USP 제어 실온 참조]. | |||
| NDC 번호 | 함량 | 설명 | 병당 정제 수 |
| 50881-005-60 | 5 mg | 한쪽 면에는 “INCY”, 다른 쪽 면에는 “5”가 새겨진 원형 정제 | 60 |
| 50881-010-60 | 10 mg | 한쪽 면에는 “INCY”, 다른 쪽 면에는 “10”이 새겨진 원형 정제 | 60 |
| 50881-015-60 | 15 mg | 한쪽 면에는 “INCY”, 다른 쪽 면에는 “15”가 새겨진 타원형 정제 | 60 |
| 50881-020-60 | 20 mg | 한쪽 면에는 “INCY”, 다른 쪽 면에는 “20”이 새겨진 캡슐형 정제 | 60 |
| 50881-025-60 | 25 mg | 한쪽 면에는 “INCY”, 다른 쪽 면에는 “25”가 새겨진 타원형 정제 | 60 |
17. 환자 상담 정보
환자에게 FDA 승인 환자 라벨링(환자 정보)을 읽도록 권장하십시오.
혈소판 감소증, 빈혈 및 호중구 감소증
Jakafi가 혈소판 감소증, 빈혈 및 호중구 감소증과 관련이 있으며 치료 전후에 전체 혈구 수를 모니터링해야 함을 환자에게 알리십시오. 출혈을 관찰하고 보고하도록 환자에게 조언하십시오 [경고 및 주의사항 (5.1) 참조].
감염
감염의 징후와 증상을 환자에게 알리고 그러한 징후와 증상이 나타나면 즉시 보고하도록 하십시오.
대상포진 및 진행성 다초점 백질뇌병증의 초기 징후와 증상에 대해 환자에게 알리고 그러한 증상이 관찰되면 의사의 조언을 구하도록 환자에게 조언하십시오 [경고 및 주의사항 (5.2) 참조].
Jakafi 치료 중단 또는 중지 후 증상 악화
치료 중단 후 골수증식성 종양의 징후와 증상이 다시 나타날 것으로 예상됨을 환자에게 알리십시오. 담당 의사와 상담 없이 Jakafi 치료를 중단하거나 중지하지 않도록 환자에게 지시하십시오 [경고 및 주의사항 (5.3) 참조].
비흑색종 피부암
Jakafi가 특정 비흑색종 피부암의 위험을 증가시킬 수 있음을 환자에게 알리십시오. 피부암을 앓았던 적이 있거나 새롭거나 변화하는 피부 병변을 관찰하는 경우 의료 서비스 제공자에게 알리도록 환자에게 조언하십시오 [경고 및 주의사항 (5.4) 참조].
지질 상승
Jakafi가 혈중 콜레스테롤을 증가시킬 수 있으며 혈중 콜레스테롤 수치를 모니터링해야 함을 환자에게 알리십시오 [경고 및 주의사항 (5.5) 참조].
주요 심혈관계 이상 사례(MACE)
심근경색, 뇌졸중 및 심혈관계 사망을 포함한 주요 심혈관계 이상 사례(MACE)가 Jakafi가 적응증이 아닌 질환인 류마티스 관절염 치료에 사용되는 다른 JAK 억제제를 사용한 임상 연구에서 보고되었음을 환자에게 알리십시오. 특히 현재 또는 과거 흡연자이거나 다른 심혈관계 위험 요인이 있는 환자에게 심혈관계 사례의 징후 및 증상 발생에 주의하도록 조언하십시오 [경고 및 주의사항 (5.6) 참조].
혈전증
Jakafi가 적응증이 아닌 질환인 류마티스 관절염 치료에 사용되는 다른 JAK 억제제를 사용한 임상 연구에서 DVT 및 PE 사례가 보고되었음을 환자에게 알리십시오. DVT 또는 PE의 징후 또는 증상이 나타나면 의료 서비스 제공자에게 알리도록 환자에게 조언하십시오 [경고 및 주의사항 (5.7) 참조].
이차 악성 종양
특히 현재 또는 과거 흡연자이고 알려진 이차 악성 종양(성공적으로 치료된 NMSC 제외)이 있는 환자에게 Jakafi가 적응증이 아닌 질환인 류마티스 관절염 치료에 사용되는 다른 JAK 억제제를 사용한 임상 연구에서 림프종 및 기타 악성 종양(NMSC 제외)이 보고되었음을 알리십시오 [경고 및 주의사항 (5.8) 참조].
약물-약물 상호작용
환자가 복용 중인 모든 약물(일반 의약품, 허브 제품 및 건강 보조 식품 포함)을 의료 서비스 제공자에게 알리도록 조언하십시오 [약물 상호작용 (7.1) 및 임상 약리학 (12.3) 참조].
투석
투석 중인 환자에게 투석 전에는 복용량을 복용하지 않고 투석 후에만 복용해야 함을 알리십시오 [용량 및 투여 (2.7) 참조].
수유
Jakafi 치료 중 및 마지막 복용 후 2주 동안 모유 수유를 하지 않도록 여성에게 알리십시오 [특정 집단에서의 사용 (8.2) 참조].
준수
의사가 지시하는 한 매일 Jakafi를 계속 복용하고 이것이 장기 치료임을 환자에게 조언하십시오. 환자는 먼저 의사와 상담하지 않고 복용량을 변경하거나 Jakafi 복용을 중단해서는 안 됩니다. 치료 중단 후 골수증식성 종양의 징후와 증상이 다시 나타날 것으로 예상됨을 환자는 알고 있어야 합니다.
제조업체:
Incyte Corporation
1801 Augustine Cut-off
Wilmington, DE 19803
Jakafi는 Incyte의 등록 상표입니다. 모든 권리는 Incyte에 있습니다.
미국 특허 번호 7598257; 8415362; 8722693; 8822481; 8829013; 9079912; 9814722; 10016429
© 2011-2023 Incyte Corporation. 모든 권리는 Incyte에 있습니다.
환자 사용 설명서
|
환자 정보 |
|
|
Jakafi란 무엇입니까? Jakafi는 다음 질환 치료에 사용되는 처방약입니다.
Jakafi가 골수섬유증 또는 진성적혈구증가증 치료에 있어 어린이에게 안전하고 효과적인지 여부는 알려져 있지 않습니다. |
|
|
Jakafi 복용 전에 다음과 같은 의학적 상태를 포함하여 의료 제공자에게 알리십시오.
처방약과 일반의약품, 비타민 및 허브 보충제를 포함하여 복용하는 모든 약에 대해 의료 제공자에게 알리십시오. 특정 다른 약물과 Jakafi를 함께 복용하면 Jakafi의 작용 방식에 영향을 미칠 수 있습니다. 복용하는 약을 알고 있어야 합니다. 새로운 약을 받을 때 의료 제공자와 약사에게 보여줄 수 있도록 약물 목록을 보관하십시오. |
|
Jakafi는 어떻게 복용해야 합니까?
|
|
|
Jakafi의 가능한 부작용은 무엇입니까? Jakafi는 다음을 포함한 심각한 부작용을 유발할 수 있습니다. 혈구 감소증. Jakafi는 혈소판 감소증(혈소판 감소증), 적혈구 감소증(빈혈) 및 백혈구 감소증(호중구 감소증)을 유발할 수 있습니다. 출혈이 발생하면 Jakafi 복용을 중단하고 의료 제공자에게 연락하십시오. 의료 제공자는 Jakafi 복용을 시작하기 전과 Jakafi 치료 중 정기적으로 혈액 검사를 실시하여 혈구 수를 확인합니다. 다음 증상 중 하나라도 발생하거나 악화되는 경우 즉시 의료 제공자에게 알리십시오. |
|
|
|
|
감염. Jakafi 치료 중에 심각한 감염이 발생할 위험이 있을 수 있습니다. 다음 감염 증상 중 하나라도 발생하는 경우 의료 제공자에게 알리십시오. |
|
|
|
|
암. Jakafi 치료 중에 일부 환자에게 비흑색종 피부암이 발생했습니다. 의료 서비스 제공자는 Jakafi 치료 기간 동안 정기적으로 피부를 검사합니다. Jakafi 치료 중 새로운 피부 병변이 발생하거나 변화가 있으면 의료 서비스 제공자에게 알리십시오.
콜레스테롤 증가. Jakafi 치료 중 혈중 콜레스테롤 수치가 변할 수 있습니다. 의료 서비스 제공자는 Jakafi 복용을 시작한 후 약 8~12주마다 필요에 따라 콜레스테롤 수치를 확인하기 위해 혈액 검사를 실시합니다.
심혈관 질환 위험 요소가 있고 현재 또는 과거 흡연자인 사람들에게서 다른 JAK 억제제를 사용하여 류마티스 관절염을 치료하는 동안 심장마비, 뇌졸중 또는 사망과 같은 주요 심혈관계 사건의 위험이 증가합니다.
Jakafi 복용 중 심장마비 또는 뇌졸중 증상이 나타나면 즉시 응급 치료를 받으십시오. 증상은 다음과 같습니다.
혈전 위험 증가. 다리 정맥(심부 정맥 혈전증, DVT) 또는 폐(폐색전증, PE)의 혈전이 류마티스 관절염에 대한 다른 JAK 억제제를 복용하는 사람들에게 발생했으며 생명을 위협할 수 있습니다.
특정 유형의 MF 및 PV가 있는 성인에서 Jakafi의 가장 흔한 부작용은 다음과 같습니다. |
|
|
|
|
aGVHD 환자에서 Jakafi의 가장 흔한 부작용은 다음과 같습니다. |
|
|
|
| cGVHD 환자에서 Jakafi의 가장 흔한 부작용은 다음과 같습니다. |
|
|
|
| 이것들은 Jakafi의 모든 가능한 부작용이 아닙니다. 부작용에 대한 의학적 조언은 의사에게 문의하십시오. FDA에 부작용을 보고할 수 있습니다(1-800-FDA-1088). Incyte Corporation에도 부작용을 보고할 수 있습니다(1-855-463-3463). |
|
|
Jakafi는 어떻게 보관해야 합니까?
Jakafi 및 모든 의약품을 어린이의 손이 닿지 않는 곳에 보관하십시오. |
|
| Jakafi의 안전하고 효과적인 사용에 대한 일반 정보.
의약품은 환자 정보에 나열된 목적 이외의 목적으로 처방되는 경우가 있습니다. 처방되지 않은 질환에 Jakafi를 사용하지 마십시오. 동일한 증상이 있는 다른 사람에게 Jakafi를 제공하지 마십시오. 해를 끼칠 수 있습니다. 약사 또는 의료 서비스 제공자에게 의료 전문가를 위해 작성된 정보를 요청할 수 있습니다. |
|
|
Jakafi의 성분은 무엇입니까? 비활성 성분: 미정질 셀룰로오스, 유당 일수화물, 마그네슘 스테아레이트, 콜로이드성 이산화규소, 나트륨 전분 글리콜레이트, 포비돈 및 히드록시프로필 셀룰로오스 제조사: Incyte Corporation, 1801 Augustine Cut-off, Wilmington, DE 19803 Jakafi는 Incyte의 등록 상표입니다. 모든 권리는 보호됩니다. © 2011-2021 Incyte Corporation. 모든 권리는 보호됩니다. |
|
이 환자 정보는 미국 식품의약국(FDA)의 승인을 받았습니다. 개정: 2021년 9월
5mg 정제 병 라벨
Rx only
NDC 50881-005-60
Jakafi® (룩소리티닙) 정제
5 mg
60정
각 정제는 룩소리티닙(ruxolitinib) 유리염기 5mg에 해당하는 룩소리티닙(ruxolitinib) 인산염 6.6mg을 함유합니다.

10mg 정제 병 라벨
Rx only
NDC 50881-010-60
Jakafi® (룩소리티닙) 정제
10 mg
60정
각 정제는 13.2 mg의 ruxolitinib phosphate를 함유하고 있으며, 이는 10 mg의 ruxolitinib free base에 해당합니다.

15mg 정제 병 라벨
처방전 의약품
NDC 50881-015-60
Jakafi® (Ruxolitinib) 정제
15 mg
60정
각 정제는 15 mg의 ruxolitinib free base에 해당하는 19.8 mg의 ruxolitinib phosphate를 함유하고 있습니다.

20mg 정제 병 라벨
처방전 의약품
NDC 50881-020-60
Jakafi® (Ruxolitinib) 정
20 mg
60정
각 정제는 20 mg의 ruxolitinib free base에 해당하는 26.4 mg의 ruxolitinib phosphate를 함유하고 있습니다.

25mg 정제 병 라벨
처방전 의약품
NDC 50881-025-60
Jakafi® (Ruxolitinib) 정제
25 mg
60정
각 정제는 25 mg ruxolitinib free base에 해당하는 33 mg ruxolitinib phosphate를 함유하고 있습니다.
