
의약품 제조업체: Cordavis Limited (Updated: 2023-11-16)
처방 정보 요약
HUMIRA® (adalimumab) injection, for subcutaneous use
미국 최초 승인: 2002
경고: 심각한 감염 및 악성 종양
전체 경고 상자에 대한 전체 처방 정보를 참조하십시오.
-
결핵(TB), 세균성 패혈증, 침윤성 진균 감염(히스토플라스마증 등) 및 기타 기회 감염으로 인한 감염을 포함하여 입원 또는 사망으로 이어지는 심각한 감염 위험 증가.
-
치료 중 환자가 심각한 감염이나 패혈증을 앓는 경우 HUMIRA를 중단하십시오.
-
잠복성 결핵 검사를 실시하십시오. 양성인 경우 HUMIRA를 시작하기 전에 결핵 치료를 시작하십시오.
- 초기 잠복성 결핵 검사가 음성이더라도 치료 중 모든 환자의 활동성 결핵을 모니터링하십시오.
악성 종양 (5.2):
-
HUMIRA를 포함한 TNF 억제제로 치료받은 소아 및 청소년 환자에서 치명적인 경우도 있는 림프종 및 기타 악성 종양이 보고되었습니다.
- HUMIRA를 포함한 TNF 억제제로 치료받은 염증성 장 질환이 있는 청소년 및 젊은 성인에서 드문 유형의 T 세포 림프종인 간비장 T 세포 림프종(HSTCL)의 시판 후 사례가 발생했습니다.
적응증 및 사용
HUMIRA는 종양 괴사 인자(TNF) 억제제로 다음에 사용됩니다.
- 류마티스 관절염(RA) (1.1): 중등도에서 중증의 활동성 RA가 있는 성인 환자에서 징후 및 증상을 감소시키고, 주요 임상 반응을 유도하고, 구조적 손상의 진행을 억제하고, 신체 기능을 개선합니다.
- 소아 특발성 관절염(JIA) (1.2): 2세 이상의 환자에서 중등도에서 중증의 활동성 다관절 JIA의 징후 및 증상을 감소시킵니다.
- 건선성 관절염(PsA) (1.3): 활동성 PsA가 있는 성인 환자에서 징후 및 증상을 감소시키고, 구조적 손상의 진행을 억제하고, 신체 기능을 개선합니다.
- 강직성 척추염(AS) (1.4): 활동성 AS가 있는 성인 환자에서 징후 및 증상을 감소시킵니다.
- 크론병(CD) (1.5): 성인 및 6세 이상의 소아 환자에서 중등도에서 중증의 활동성 크론병 치료.
-
궤양성 대장염(UC) (1.6): 성인 및 5세 이상의 소아 환자에서 중등도에서 중증의 활동성 궤양성 대장염 치료.
사용 제한: TNF 억제제에 대한 반응이 없거나 내약성이 없는 환자에서는 효능이 입증되지 않았습니다. - 판상 건선(Ps) (1.7): 전신 요법 또는 광선 요법의 대상이 되는 중등도에서 중증의 만성 판상 건선이 있는 성인 환자 치료. 다른 전신 요법이 의학적으로 적합하지 않은 경우.
- 농포성 한선염(HS) (1.8): 12세 이상의 환자에서 중등도에서 중증의 농포성 한선염 치료.
- 포도막염(UV) (1.9): 성인 및 2세 이상의 소아 환자에서 비감염성 중간, 후방 및 전포도막염 치료.
투여량 및 투여 방법
- 피하 주사로 투여 (2)
류마티스 관절염, 건선성 관절염, 강직성 척추염 (2.1):
-
성인: 격주로 40mg.
- 메토트렉세이트를 투여받지 않는 일부 RA 환자는 투여량을 매주 40mg 또는 격주로 80mg으로 증가시키면 도움이 될 수 있습니다.
소아 특발성 관절염 또는 소아 포도막염 (2.2):
| 소아 체중 2세 이상 |
권장 투여량 |
| 10kg(22lbs)에서 15kg(33lbs) 미만 | 격주로 10mg |
| 15kg(33lbs)에서 30kg(66lbs) 미만 | 격주로 20mg |
| 30kg(66lbs) 이상 | 격주로 40mg |
크론병 (2.3):
- 성인: 1일차에 160mg (하루에 투여하거나 이틀 연속으로 나눠 투여); 15일차에 80mg; 29일차부터 격주로 40mg 투여.
- 6세 이상 소아 환자:
| 소아 체중 |
권장 용량 | |
| 1일차 및 15일차 | 29일차부터 | |
| 17kg (37lbs) 미만 40kg (88lbs) |
1일차: 80mg 15일차: 40mg |
격주로 20mg |
| 40kg (88lbs) 이상 |
1일차: 160mg (단일 용량 또는 이틀 연속으로 나눠 투여) 15일차: 80mg |
격주로 40mg |
궤양성 대장염 (2.4):
- 성인: 1일차에 160mg (하루에 투여하거나 이틀 연속으로 나눠 투여), 15일차에 80mg, 29일차부터 격주로 40mg 투여. 8주 (57일차)까지 임상적 관해 증거가 없는 환자는 투약을 중단합니다.
- 5세 이상 소아 환자:
| 소아 체중 |
권장 용량 | |
| 1일차부터 15일차까지 | 29일차부터* | |
| 20kg (44lbs) 미만 40kg (88lbs) |
1일차: 80mg 8일차: 40mg 15일차: 40mg |
격주로 40mg 또는 매주 20mg |
| 40kg (88lbs) 이상 | 1일차: 160mg (단일 용량 또는 이틀 연속으로 나눠 투여) 8일차: 80mg 15일차: 80mg |
격주로 80mg 또는 매주 40mg |
| * 18세가 되고 HUMIRA 요법으로 잘 조절되는 환자는 권장 소아 용량을 계속 투여합니다. | ||
판상 건선 또는 성인 포도막염 (2.5):
- 성인: 80mg 초기 용량을 투여한 후, 초기 용량 투여 후 1주일 후부터 격주로 40mg 투여.
털 난 땀샘 염 (2.6):
-
성인:
◦ 1일차: 160mg (하루에 투여하거나 이틀 연속으로 나눠 투여)
◦ 15일차: 80mg
◦ 29일차 이후 용량: 매주 40mg 또는 격주로 80mg - 12세 이상 청소년:
| 청소년 체중 |
권장 용량 |
| 30kg (66lbs) 미만 60kg (132lbs) |
1일차: 80mg 8일차 이후 용량: 격주로 40mg |
| 60kg (132lbs) 이상 |
1일차: 160mg (하루에 투여하거나 이틀 연속으로 나눠 투여) 15일차: 80mg 29일차 이후 용량: 매주 40mg 또는 격주로 80mg |
투약 형태 및 강도
금기 사항
없음 (4)
경고 및 주의 사항
- 심각한 감염: 활동적인 감염 중에 HUMIRA를 시작하지 마십시오. 감염이 발생하면 주의 깊게 모니터링하고 감염이 심각해지면 HUMIRA를 중단하십시오. (5.1)
- 침윤성 진균 감염: HUMIRA를 복용하는 동안 전신 질환이 발생하는 환자의 경우, 진균증이 풍토병인 지역에 거주하거나 여행하는 환자에게는 경험적 항진균 요법을 고려하십시오. (5.1)
- 악성 종양: HUMIRA를 투여받은 환자의 악성 종양 발생률은 대조군보다 높았습니다. (5.2)
- 아나필락시스 또는 심각한 과민 반응이 발생할 수 있습니다. (5.3)
- B형 간염 바이러스 재활성화: 치료 중 및 치료 후 몇 달 동안 HBV 보균자를 모니터링하십시오. 재활성화가 발생하면 HUMIRA를 중단하고 항바이러스 치료를 시작하십시오. (5.4)
- 탈수초 질환: 악화 또는 새롭게 발생할 수 있습니다. (5.5)
- 혈구 감소증, 범혈구 감소증: 증상이 나타나면 즉시 의사의 진료를 받도록 환자에게 조언하고 HUMIRA를 중단하는 것을 고려하십시오. (5.6)
- 심부전: 악화 또는 새롭게 발생할 수 있습니다. (5.8)
- 루푸스 유사 증후군: 증후군이 발생하면 HUMIRA를 중단하십시오. (5.9)
부작용
가장 흔한 부작용 (>10%)은 감염 (예: 상기도 감염, 부비강염), 주사 부위 반응, 두통 및 발진입니다. (6.1)
의심되는 부작용을 보고하려면 AbbVie Inc.에 1-800-633-9110 또는 FDA에 1-800-FDA-1088 또는 www.fda.gov/medwatch로 연락하십시오.
약물 상호 작용
환자 상담 정보 및 약물 안내서를 보려면 17을 참조하십시오.
개정: 2023년 11월
목차
전문 정보: 내용*
경고: 심각한 감염 및 악성 종양
1 적응증 및 사용법
1.1 류마티스 관절염
1.2 소아 특발성 관절염
1.3 건선성 관절염
1.4 강직성 척추염
1.5 크론병
1.6 궤양성 대장염
1.7 판상 건선
1.8 땀샘 농피증
1.9 포도막염
2 용법 및 용량
2.1 류마티스 관절염, 건선성 관절염 및 강직성 척추염
2.2 소아 특발성 관절염 또는 소아 포도막염
2.3 크론병
2.4 궤양성 대장염
2.5 판상 건선 또는 성인 포도막염
2.6 땀샘 농피증
2.7 안전성 평가를 위한 모니터링
2.8 투여에 대한 일반적인 고려 사항
3 용법 및 용량
4 금기 사항
5 경고 및 주의 사항
5.1 심각한 감염
5.2 악성 종양
5.3 과민 반응
5.4 B형 간염 바이러스 재활성화
5.5 신경계 반응
5.6 혈액학적 반응
5.7 아나킨라와 함께 사용할 경우 감염 위험 증가
5.8 심부전
5.9 자가 면역
5.10 예방 접종
5.11 아바타셉트와 함께 사용할 경우 감염 위험 증가
6 이상 반응
6.1 임상 시험 경험
6.2 면역원성
6.3 시판 후 경험
7 약물 상호 작용
7.1 메토트렉세이트
7.2 생물학적 제제
7.3 생백신
7.4 시토크롬 P450 기질
8 특정 인구 집단에서의 사용
8.1 임신
8.2 수유
8.4 소아 사용
8.5 노인 사용
10 과량 투여
11 설명
12 임상 약리
12.1 작용 기전
12.2 약력학
12.3 약동학
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식 능력 저해
14 임상 연구
14.1 류마티스 관절염
14.2 소아 특발성 관절염
14.3 건선성 관절염
14.4 강직성 척추염
14.5 성인 크론병
14.6 소아 크론병
14.7 성인 궤양성 대장염
14.8 소아 궤양성 대장염
14.9 판상 건선
14.10 땀샘 농피증
14.11 성인 포도막염
14.12 소아 포도막염
15 참고 문헌
16 포장 단위/보관 및 취급
17 환자 상담 정보
- *
- 전문 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
경고 사항
경고: 심각한 감염 및 악성 종양
심각한 감염
HUMIRA로 치료받는 환자는 입원 또는 사망으로 이어질 수 있는 심각한 감염 위험이 증가합니다. [경고 및 주의 사항 (5.1) 참조]. 이러한 감염이 발생한 대부분의 환자는 메토트렉세이트 또는 코르티코스테로이드와 같은 면역 억제제를 병용했습니다.
환자가 심각한 감염이나 패혈증을 앓는 경우 HUMIRA를 중단하십시오.
보고된 감염에는 다음이 포함됩니다.
-
잠복성 결핵의 재활성화를 포함한 활동성 결핵(TB). 결핵 환자는 종종 전신성 또는 폐외 질환으로 나타났습니다. HUMIRA 사용 전과 치료 중에 환자를 결핵 잠복 감염에 대해 검사하십시오. HUMIRA 사용 전에 잠복성 결핵 치료를 시작하십시오.
-
히스토플라스마증, 콕시디오이데스 진균증, 칸디다증, 아스페르길루스증, 블라스토미세스증 및 폐렴구균증을 포함한 침윤성 진균 감염. 히스토플라스마증 또는 기타 침윤성 진균 감염이 있는 환자는 국소화된 질환이 아닌 전신성 질환으로 나타날 수 있습니다. 히스토플라스마증에 대한 항원 및 항체 검사는 활동성 감염이 있는 일부 환자에서 음성일 수 있습니다. 심각한 전신 질환이 발생하는 침윤성 진균 감염 위험이 있는 환자에게는 경험적 항진균 치료를 고려하십시오.
- 레지오넬라 및 리스테리아를 포함한 기회 감염균으로 인한 박테리아, 바이러스 및 기타 감염.
만성 또는 재발성 감염이 있는 환자에서 치료를 시작하기 전에 HUMIRA 치료의 위험과 이점을 신중하게 고려하십시오.
HUMIRA 치료 중 및 후에 감염의 징후와 증상, 치료 시작 전에 잠복성 결핵 감염 검사에서 음성으로 판정된 환자에서 결핵 발생 가능성을 포함하여 환자를 면밀히 모니터링하십시오. [경고 및 주의 사항 (5.1) 및 이상 반응 (6.1) 참조].
악성 종양
HUMIRA를 포함한 TNF 억제제로 치료받은 소아 및 청소년 환자에서 일부는 치명적인 림프종 및 기타 악성 종양이 보고되었습니다. [경고 및 주의 사항 (5.2) 참조]. HUMIRA를 포함한 TNF 억제제로 치료받은 환자에서 드문 유형의 T 세포 림프종인 간비장 T 세포 림프종(HSTCL)의 시판 후 사례가 보고되었습니다. 이러한 사례는 매우 공격적인 질병 경과를 보였으며 치명적이었습니다. 보고된 TNF 억제제 사례의 대부분은 크론병 또는 궤양성 대장염 환자였으며 대부분은 청소년 및 젊은 성인 남성이었습니다. 이러한 환자의 거의 대부분은 진단 시 또는 그 이전에 TNF 억제제와 함께 아자티오프린 또는 6-머캅토퓨린(6-MP)으로 치료를 받았습니다. HSTCL의 발생이 TNF 억제제 사용과 관련이 있는지 또는 이러한 다른 면역 억제제와 함께 TNF 억제제를 사용한 것과 관련이 있는지는 불확실합니다. [경고 및 주의 사항 (5.2) 참조].
1 적응증 및 용법
1.1 류마티스 관절염
HUMIRA는 중등도에서 중증의 활동성 류마티스 관절염 성인 환자에서 징후 및 증상을 감소시키고, 주요 임상 반응을 유도하고, 구조적 손상의 진행을 억제하고, 신체 기능을 개선하는 데 사용됩니다. HUMIRA는 단독으로 또는 메토트렉세이트 또는 다른 비생물학적 질병 수정 항류마티스 약물(DMARDs)과 병용하여 사용할 수 있습니다.
1.2 소아 특발성 관절염
HUMIRA는 2세 이상의 중등도에서 중증의 활동성 다관절 소아 특발성 관절염 환자에서 징후 및 증상을 감소시키는 데 사용됩니다. HUMIRA는 단독으로 또는 메토트렉세이트와 병용하여 사용할 수 있습니다.
1.3 건선성 관절염
HUMIRA는 활동성 건선성 관절염 성인 환자에서 징후 및 증상을 감소시키고, 구조적 손상의 진행을 억제하고, 신체 기능을 개선하는 데 사용됩니다. HUMIRA는 단독으로 또는 비생물학적 DMARDs와 병용하여 사용할 수 있습니다.
1.6 궤양성 대장염
HUMIRA는 성인 및 5세 이상의 소아 환자에서 중등도에서 중증의 활동성 궤양성 대장염 치료에 사용됩니다.
사용 제한
HUMIRA의 효능은 TNF 억제제에 대한 반응이 없거나 내약성이 없는 환자에서는 확립되지 않았습니다. [임상 연구 (14.7, 14.8)].
1.7 판상 건선
HUMIRA는 전신 요법 또는 광선 요법의 대상이 되는 중등도에서 중증의 만성 판상 건선 성인 환자에서, 다른 전신 요법이 의학적으로 적합하지 않을 때 사용됩니다. HUMIRA는 의사의 면밀한 모니터링을 받고 정기적으로 추적 검사를 받는 환자에게만 투여해야 합니다. [경고 및 주의 사항 (5)].
2. 용법용량
2.1 류마티스 관절염, 건선성 관절염 및 강직성 척추염
류마티스 관절염(RA), 건선성 관절염(PsA) 또는 강직성 척추염(AS) 성인 환자의 경우 HUMIRA의 권장 피하 투여량은 격주로 40mg입니다. 메토트렉세이트(MTX), 기타 비생물학적 DMARDS, 글루코코르티코이드, 비스테로이드성 항염증제(NSAIDs) 및/또는 진통제는 HUMIRA 치료 중에 계속 사용할 수 있습니다. RA 치료에서 MTX를 동시에 복용하지 않는 일부 환자는 HUMIRA의 투여량을 매주 40mg 또는 격주로 80mg으로 증가시키면 추가적인 이점을 얻을 수 있습니다.
2.2 소아 특발성 관절염 또는 소아 포도막염
다관절 소아 특발성 관절염(JIA) 또는 소아 포도막염이 있는 2세 이상 환자의 경우 HUMIRA의 권장 피하 투여량은 아래와 같이 체중을 기준으로 합니다. MTX, 글루코코르티코이드, NSAIDs 및/또는 진통제는 HUMIRA 치료 중에 계속 사용할 수 있습니다.
| 소아 체중 (2세 이상) |
권장 투여량 |
| 10kg(22lbs)에서 15kg(33lbs) 미만 | 격주로 10mg |
| 15kg(33lbs)에서 30kg(66lbs) 미만 | 격주로 20mg |
| 30kg(66lbs) 이상 | 격주로 40mg |
HUMIRA는 2세 미만의 다관절 JIA 또는 소아 포도막염 환자 또는 체중이 10kg 미만인 환자에게 연구되지 않았습니다.
2.3 크론병
성인
크론병(CD) 성인 환자의 경우 HUMIRA의 권장 피하 투여량은 1일차에 160mg(하루에 투여하거나 연이틀에 나누어 투여)으로 시작하여 2주 후(15일차)에 80mg을 투여합니다. 2주 후(29일차)에 격주로 40mg 투여를 시작합니다. 아미노살리실레이트 및/또는 코르티코스테로이드는 HUMIRA 치료 중에 계속 사용할 수 있습니다. 아자티오프린, 6-머캅토퓨린(6-MP) [경고 및 주의 사항 (5.2)] 또는 MTX는 필요한 경우 HUMIRA 치료 중에 계속 사용할 수 있습니다.
소아
크론병(CD)이 있는 6세 이상 소아 환자의 경우 HUMIRA의 권장 피하 투여량은 아래와 같이 체중을 기준으로 합니다.
| 소아 체중 |
권장 투여량 | |
| 1일부터 15일까지 | 29일부터 시작 | |
| 17kg(37lbs)에서 40kg(88lbs) 미만 |
1일차: 80mg 15일차: 40mg |
격주로 20mg |
| 40kg(88lbs) 이상 | 1일차: 160mg(단일 투여 또는 연이틀에 나누어 투여) 15일차: 80mg |
격주로 40mg |
2.4 궤양성 대장염
성인
궤양성 대장염 성인 환자의 경우 HUMIRA의 권장 피하 투여량은 1일차에 160mg(하루에 투여하거나 이틀 연속으로 나눠 투여)을 처음 투여한 후 2주 후(15일차)에 80mg을 투여합니다. 2주 후(29일차)부터는 격주로 40mg을 투여합니다.
8주(57일차) 치료 후 임상적 관해가 나타나지 않는 성인 환자의 경우 HUMIRA 투여를 중단합니다. HUMIRA 치료 중에 아미노살리실산염 및/또는 코르티코스테로이드를 계속 투여할 수 있습니다. 필요한 경우 아자티오프린 및 6-머캅토퓨린(6-MP) [경고 및 주의 사항 (5.2) 참조]을 HUMIRA 치료 중에 계속 투여할 수 있습니다.
소아
궤양성 대장염이 있는 5세 이상 소아 환자의 경우 HUMIRA의 권장 피하 투여량은 아래와 같이 체중에 따라 결정됩니다.
| 소아 체중 | 권장 투여량 | |
| 1일부터 15일까지 | 29일차부터* | |
| 20kg(44파운드)에서 40kg(88파운드) 미만 |
1일차: 80mg 8일차: 40mg 15일차: 40mg |
격주로 40mg 또는 매주 20mg |
| 40kg(88파운드) 이상 | 1일차: 160mg(단일 투여 또는 이틀 연속으로 나눠 투여) 8일차: 80mg 15일차: 80mg |
격주로 80mg 또는 매주 40mg |
| * 18세가 되고 HUMIRA 요법으로 잘 조절되는 환자의 경우 권장 소아 투여량을 계속 투여합니다. | ||
2.5 판상 건선 또는 성인 포도막염
판상 건선(Ps) 또는 포도막염(UV) 성인 환자의 경우 HUMIRA의 권장 피하 투여량은 80mg을 처음 투여한 후 1주일 후부터 격주로 40mg을 투여합니다. 1년 이상 중등도에서 중증의 만성 Ps에 대한 HUMIRA의 사용은 대조군 임상 연구에서 평가되지 않았습니다.
2.6 털샘 염
성인
털샘 염(HS) 성인 환자의 경우 HUMIRA의 권장 피하 투여량은 160mg(하루에 투여하거나 이틀 연속으로 나눠 투여)을 처음 투여한 후 2주 후(15일차)에 80mg을 투여합니다. 2주 후(29일차)부터는 매주 40mg 또는 격주로 80mg을 투여합니다.
청소년
털샘 염(HS)이 있는 체중이 30kg 이상인 12세 이상 청소년 환자의 경우 HUMIRA의 권장 피하 투여량은 아래와 같이 체중에 따라 결정됩니다. [특정 인구 집단에서의 사용 (8.4) 및 임상 약리학 (12.3) 참조]:
| 청소년 환자의 체중 (12세 이상) |
권장 투여량 |
| 30kg(66파운드)에서 60kg(132파운드) 미만 |
|
| 60kg(132파운드) 이상 |
|
2.8 투여에 대한 일반적인 고려 사항
HUMIRA는 의사의 지시와 감독 하에 사용하도록 되어 있습니다. 환자는 HUMIRA를 스스로 주사하거나, 의사가 적절하다고 판단하는 경우, 보호자가 HUMIRA Pen 또는 미리 채워진 주사기를 사용하여 HUMIRA를 주사할 수 있습니다. 피하 주사 기술에 대한 적절한 교육을 받은 후 필요에 따라 의료적 추적 관찰을 받아야 합니다.
HUMIRA는 액체가 실온에 도달하도록 주사하기 전에 15~30분 동안 냉장고에서 꺼낼 수 있습니다. 실온에 도달하도록 할 때 뚜껑이나 덮개를 제거하지 마십시오. 피하 투여 전에 HUMIRA Pen, 미리 채워진 주사기 또는 단회용 기관 사용 바이알의 용액을 침전물과 변색 여부를 주의 깊게 검사하십시오. 침전물과 변색이 발견되면 제품을 사용하지 마십시오. HUMIRA는 방부제를 함유하지 않습니다. 따라서 주사기에서 남은 약물의 사용하지 않은 부분은 버리십시오. 참고: 라텍스에 민감한 환자는 HUMIRA 40 mg/0.8 mL Pen 및 40 mg/0.8 mL, 20 mg/0.4 mL 및 10 mg/0.2 mL 미리 채워진 주사기의 바늘 덮개를 만지지 않도록 지시하십시오. 이는 천연 고무 라텍스를 함유할 수 있기 때문입니다 [제품 정보/보관 및 취급 (16)].
HUMIRA Pen 또는 미리 채워진 주사기를 사용하는 환자에게는 사용 설명서에 제공된 지침에 따라 주사기에 있는 용액을 모두 주사하도록 지시하십시오 [ 사용 설명서].
주사는 허벅지 또는 복부의 별도 부위에서 이루어져야 합니다. 주사 부위를 바꿔 주고 피부가 부드럽지 않거나 멍이 들거나 붉거나 단단한 부위에는 주사하지 마십시오.
복용량을 놓친 경우 가능한 한 빨리 복용량을 투여하십시오. 그 후에는 정기적으로 예정된 시간에 복용을 재개하십시오.
HUMIRA 단회용 기관 사용 바이알은 병원, 의사 사무실 또는 클리닉과 같은 기관 환경에서만 투여하도록 되어 있습니다. 멸균 바늘과 주사기를 사용하여 용량을 뽑아내고 기관 환경에서 의료 서비스 제공자가 즉시 투여하십시오. 바이알당 한 번만 투여하십시오. 바이알은 방부제를 함유하지 않습니다. 따라서 사용하지 않은 부분은 버리십시오.
3가지 용량 형태 및 함량
HUMIRA는 다음과 같은 무색의 투명한 용액입니다.
-
펜 (HUMIRA Pen)
주사제: 80 mg/0.8 mL, 일회용 펜
주사제: 40 mg/0.8 mL, 일회용 펜
주사제: 40 mg/0.4 mL, 일회용 펜 -
사전 충전형 주사기
주사제: 80 mg/0.8 mL, 일회용 사전 충전형 유리 주사기
주사제: 40 mg/0.8 mL, 일회용 사전 충전형 유리 주사기
주사제: 40 mg/0.4 mL, 일회용 사전 충전형 유리 주사기
주사제: 20 mg/0.4 mL, 일회용 사전 충전형 유리 주사기
주사제: 20 mg/0.2 mL, 일회용 사전 충전형 유리 주사기
주사제: 10 mg/0.2 mL, 일회용 사전 충전형 유리 주사기
주사제: 10 mg/0.1 mL, 일회용 사전 충전형 유리 주사기 -
일회용 기관용 바이알
주사제: 40 mg/0.8 mL, 기관용 일회용 유리 바이알
4 금기 사항
없음.
경고 및 주의사항 5가지
5.1 심각한 감염
HUMIRA로 치료받는 환자는 다양한 기관계 및 부위에 영향을 미치는 심각한 감염 위험이 증가하며, 이는 입원 또는 사망으로 이어질 수 있습니다. 아스페르길루스증, 블라스토미코스증, 칸디다증, 콕시디오이데스증, 히스토플라스마증, 레지오넬라증, 리스테리아증, 폐포자충증 및 결핵을 포함한 박테리아, 마이코박테리아, 침윤성 진균, 바이러스, 기생충 또는 기타 기회 감염균에 의한 기회 감염이 TNF 억제제에서 보고되었습니다. 환자는 국소화된 질환보다는 흔히 전신 질환을 보였습니다.
TNF 억제제와 아바타셉트 또는 아나킨라의 병용은 류마티스 관절염(RA) 환자에서 심각한 감염 위험이 높았습니다. 따라서 HUMIRA와 이러한 생물학적 제제의 병용은 RA 환자 치료에 권장되지 않습니다 [경고 및 주의 사항 (5.7, 5.11) 및 약물 상호 작용 (7.2)].
HUMIRA 치료는 국소 감염을 포함한 활성 감염 환자에게 시작해서는 안 됩니다. 65세 이상 환자, 동반 질환이 있는 환자 및/또는 면역 억제제(예: 코르티코스테로이드 또는 메토트렉세이트)를 병용하는 환자는 감염 위험이 더 높을 수 있습니다. 다음 환자에서 치료를 시작하기 전에 치료의 위험과 이점을 고려하십시오.
- 만성 또는 재발성 감염이 있는 환자;
- 결핵에 노출된 환자;
- 기회 감염 병력이 있는 환자;
- 결핵 또는 히스토플라스마증, 콕시디오이데스증 또는 블라스토미코스증과 같은 풍토병성 진균증이 유행하는 지역에 거주하거나 여행한 환자; 또는
- 감염에 취약하게 만드는 기저 질환이 있는 환자.
결핵
잠복 또는 활성 결핵 치료를 이전에 받은 환자를 포함하여 HUMIRA를 투여받은 환자에서 결핵 재활성화 및 새로 발생한 결핵 감염 사례가 보고되었습니다. 보고된 사례에는 폐 및 폐 외(즉, 전신) 결핵이 포함되었습니다. HUMIRA를 시작하기 전과 치료 중 주기적으로 환자의 결핵 위험 요인을 평가하고 잠복 감염을 검사하십시오.
TNF 차단제 치료 전에 잠복 결핵 감염을 치료하면 치료 중 결핵 재활성화 위험을 줄이는 것으로 나타났습니다. HUMIRA를 시작하기 전에 잠복 결핵 치료가 필요한지 평가하십시오. 이전에 Bacille Calmette-Guerin(BCG)으로 예방 접종을 받은 환자의 경우에도 ≥ 5mm의 경결을 양성 결핵 피부 반응 결과로 간주하십시오.
적절한 치료 과정을 확인할 수 없는 과거 잠복 또는 활성 결핵 병력이 있는 환자와 잠복 결핵 검사는 음성이지만 결핵 감염 위험 요인이 있는 환자의 경우 HUMIRA를 시작하기 전에 항결핵 요법을 고려하십시오. 결핵 예방 치료에도 불구하고 HUMIRA로 치료받는 환자에서 결핵이 재활성화된 사례가 발생했습니다. 개별 환자에게 항결핵 요법 시작이 적절한지 여부를 결정하는 데 도움이 되도록 결핵 치료 전문의와 상담하는 것이 좋습니다.
HUMIRA 치료 중 새로운 감염이 발생하는 환자, 특히 이전에 또는 최근에 결핵 유병률이 높은 국가를 여행했거나 활성 결핵 환자와 밀접한 접촉이 있었던 환자의 경우 결핵을 감별 진단에서 강력하게 고려하십시오.
모니터링
HUMIRA 치료 중 및 후에 감염의 징후와 증상, 특히 치료를 시작하기 전에 잠복 결핵 감염 검사에서 음성으로 판정된 환자에서 결핵이 발생하는지 면밀히 모니터링하십시오. 잠복 결핵 감염 검사는 HUMIRA 치료 중에 위음성을 보일 수도 있습니다.
환자가 심각한 감염이나 패혈증을 앓는 경우 HUMIRA를 중단하십시오. HUMIRA 치료 중에 새로운 감염이 발생하는 환자의 경우 면밀히 모니터링하고 면역 저하 환자에게 적절한 신속하고 완전한 진단 검사를 수행하고 적절한 항균 요법을 시작하십시오.
침윤성 진균 감염
환자가 심각한 전신 질환을 앓고 있으며 진균증이 유행하는 지역에 거주하거나 여행하는 경우 감별 진단에서 침윤성 진균 감염을 고려하십시오. 히스토플라스마증에 대한 항원 및 항체 검사는 활성 감염이 있는 일부 환자에서 음성일 수 있습니다. 진단 검사를 수행하는 동안 심각한 진균 감염 위험과 항진균 요법의 위험을 모두 고려하여 적절한 경험적 항진균 요법을 고려하십시오. 이러한 환자의 관리를 돕기 위해 침윤성 진균 감염 진단 및 치료 전문의와 상담하는 것을 고려하십시오.
5.2 악성 종양
성공적으로 치료된 비흑색종 피부암(NMSC)을 제외한 알려진 악성 종양이 있는 환자에서 HUMIRA를 포함한 TNF 억제제 치료의 위험과 이점을 치료를 시작하기 전에 고려하십시오. 또한 악성 종양이 발생한 환자에서 TNF 억제제를 계속 사용할지 여부를 고려할 때도 위험과 이점을 고려하십시오.
성인의 악성 종양
HUMIRA를 포함한 일부 TNF 억제제의 임상 시험 통제 그룹에서 TNF 억제제 치료를 받은 성인 환자에서 대조군 성인 환자에 비해 악성 종양 발생률이 더 높았습니다. 류마티스 관절염(RA), 건선성 관절염(PsA), 강직성 척추염(AS), 크론병(CD), 궤양성 대장염(UC), 판상 건선(Ps), 땀샘 염증(HS) 및 포도막염(UV) 성인 환자를 대상으로 한 39건의 글로벌 HUMIRA 임상 시험의 통제 그룹에서 비흑색종(기저 세포 및 편평 세포) 피부암을 제외한 악성 종양이 HUMIRA 치료를 받은 환자 7973명에서 100 환자-년당 0.7(0.48, 1.03)의 비율(95% 신뢰 구간)로 관찰된 반면, 대조군 치료를 받은 환자 4848명에서는 100 환자-년당 0.7(0.41, 1.17)의 비율로 관찰되었습니다(HUMIRA 치료를 받은 환자의 중간 치료 기간은 4개월, 대조군 치료를 받은 환자의 중간 치료 기간은 4개월). RA, PsA, AS, CD, UC, Ps, HS 및 UV 성인 환자를 대상으로 한 52건의 글로벌 통제 및 비통제 HUMIRA 임상 시험에서 림프종 및 NMSC를 제외하고 가장 흔하게 관찰된 악성 종양은 유방암, 대장암, 전립선암, 폐암 및 흑색종이었습니다. 연구의 통제 및 비통제 그룹에서 HUMIRA 치료를 받은 환자의 악성 종양은 SEER 데이터베이스(나이, 성별 및 인종 조정)에 따른 미국 일반 인구에서 예상되는 것과 유형 및 수가 유사했습니다.1
악성 종양 위험이 높은 성인 환자(즉, 흡연력이 심한 만성 폐쇄성 폐 질환 환자 및 싸이클로포스파마이드 치료를 받은 베게너 육아종증 환자)를 대상으로 한 다른 TNF 억제제의 통제 임상 시험에서 대조군에 비해 TNF 억제제 그룹에서 악성 종양이 더 많이 발생했습니다.
비흑색종 피부암
RA, PsA, AS, CD, UC, Ps, HS 및 UV 성인 환자를 대상으로 한 39건의 글로벌 HUMIRA 임상 시험의 통제 그룹에서 NMSC 발생률(95% 신뢰 구간)은 HUMIRA 치료를 받은 환자에서 100 환자-년당 0.8(0.52, 1.09)인 반면, 대조군 치료를 받은 환자에서는 100 환자-년당 0.2(0.10, 0.59)였습니다. 모든 환자, 특히 이전에 장기간 면역 억제제 치료를 받은 병력이 있는 환자 또는 PUVA 치료를 받은 건선 환자의 경우 HUMIRA 치료 전후로 NMSC 유무를 검사해야 합니다.
림프종 및 백혈병
성인을 대상으로 한 모든 TNF 억제제의 임상 시험 통제 그룹에서 TNF 억제제 치료를 받은 환자에서 대조군 치료를 받은 환자에 비해 림프종 발생률이 더 높았습니다. RA, PsA, AS, CD, UC, Ps, HS 및 UV 성인 환자를 대상으로 한 39건의 글로벌 HUMIRA 임상 시험의 통제 그룹에서 HUMIRA 치료를 받은 환자 7973명 중 2명에서 림프종이 발생한 반면, 대조군 치료를 받은 환자 4848명 중 1명에서 림프종이 발생했습니다. RA, PsA, AS, CD, UC, Ps, HS 및 UV 성인 환자를 대상으로 한 52건의 글로벌 통제 및 비통제 HUMIRA 임상 시험에서 중간 기간이 약 0.7년인 환자 24,605명과 HUMIRA 치료를 받은 환자-년이 40,215년 이상인 환자에서 림프종 발생률은 약 100 환자-년당 0.11이었습니다. 이는 SEER 데이터베이스(나이, 성별 및 인종 조정)에 따른 미국 일반 인구에서 예상되는 것보다 약 3배 높습니다.1 HUMIRA 임상 시험에서 림프종 발생률은 다른 TNF 억제제의 임상 시험에서 림프종 발생률과 비교할 수 없으며, 더 넓은 환자 집단에서 관찰되는 발생률을 예측할 수 없습니다. RA 및 기타 만성 염증성 질환 환자, 특히 질병 활성도가 높거나 면역 억제제 치료를 장기간 받은 환자는 TNF 억제제를 사용하지 않더라도 일반 인구보다 림프종 발생 위험이 높을 수 있습니다(최대 몇 배). RA 및 기타 적응증에서 TNF 억제제 사용과 관련하여 급성 및 만성 백혈병 발생 사례가 보고되었습니다. TNF 억제제 치료를 받지 않더라도 RA 환자는 일반 인구보다 백혈병 발생 위험이 높을 수 있습니다(약 2배).
소아 및 청소년 환자의 악성 종양
HUMIRA를 포함한 TNF 억제제 치료(치료 시작 연령 ≤ 18세)를 받은 소아, 청소년 및 젊은 성인에서 일부 치명적인 악성 종양이 보고되었습니다. 사례의 약 절반이 림프종, 특히 호지킨 림프종 및 비호지킨 림프종이었습니다. 다른 사례는 다양한 악성 종양을 나타냈으며, 면역 억제와 관련된 희귀 악성 종양과 소아 및 청소년에게서 일반적으로 관찰되지 않는 악성 종양이 포함되었습니다. 악성 종양은 중간 치료 기간이 30개월(범위 1~84개월) 후에 발생했습니다. 환자 대부분은 면역 억제제를 병용했습니다. 이러한 사례는 시판 후에 보고되었으며, 레지스트리 및 자발적인 시판 후 보고를 포함한 다양한 출처에서 파생되었습니다.
HUMIRA를 포함한 TNF 억제제로 치료받은 환자에서 희귀한 T 세포 림프종인 간비장 T 세포 림프종(HSTCL) 발생 사례가 시판 후 보고되었습니다. 이러한 사례는 매우 공격적인 질병 경과를 보였으며 치명적이었습니다. 보고된 TNF 억제제 사례 대부분은 크론병 또는 궤양성 대장염 환자에서 발생했으며, 대부분은 청소년 및 젊은 성인 남성이었습니다. 이러한 환자 거의 대부분은 진단 시 또는 진단 전에 TNF 억제제와 함께 아자티오프린 또는 6-머캅토퓨린(6-MP) 면역 억제제 치료를 받았습니다. HSTCL 발생이 TNF 억제제 사용 또는 이러한 다른 면역 억제제와 함께 TNF 억제제를 사용한 것과 관련이 있는지는 불확실합니다. 아자티오프린 또는 6-머캅토퓨린과 HUMIRA 병용 시 잠재적 위험을 신중하게 고려해야 합니다.
5.3 과민 반응
HUMIRA 투여 후 아나필락시스 및 혈관 부종이 보고되었습니다. 아나필락시스 또는 기타 심각한 알레르기 반응이 발생하면 HUMIRA 투여를 즉시 중단하고 적절한 치료를 시작해야 합니다. HUMIRA 임상 시험에서 과민 반응(예: 발진, 아나필락시스양 반응, 고정 약물 발진, 비특정 약물 반응, 두드러기)이 관찰되었습니다.
5.4 B형 간염 바이러스 재활성화
HUMIRA를 포함한 TNF 차단제의 사용은 이 바이러스의 만성 보균자인 환자에서 B형 간염 바이러스(HBV)의 재활성화 위험을 증가시킬 수 있습니다. 경우에 따라 TNF 차단제 치료와 함께 발생하는 HBV 재활성화는 치명적이었습니다. 이러한 보고의 대부분은 면역 체계를 억압하는 다른 약물을 동시에 투여받은 환자에서 발생했으며, 이는 HBV 재활성화에도 기여할 수 있습니다. TNF 차단제 치료를 시작하기 전에 HBV 감염 위험이 있는 환자를 대상으로 이전 HBV 감염 증거를 평가하십시오. HBV 보균자로 확인된 환자에게 TNF 차단제를 처방할 때는 주의하십시오. HBV 재활성화를 예방하기 위해 TNF 차단제 치료와 함께 항바이러스제로 HBV 보균자를 치료하는 것의 안전성이나 효능에 대한 적절한 데이터는 없습니다. HBV 보균자이고 TNF 차단제로 치료를 받아야 하는 환자의 경우 치료 기간과 치료 종료 후 몇 개월 동안 활동성 HBV 감염의 임상적 및 실험실적 징후가 있는지 이러한 환자를 면밀히 모니터링하십시오. HBV 재활성화가 발생한 환자의 경우 HUMIRA를 중단하고 적절한 지지 요법과 함께 효과적인 항바이러스 요법을 시작하십시오. HBV 재활성화가 통제된 후 TNF 차단제 치료를 재개하는 것의 안전성은 알려져 있지 않습니다. 따라서 이러한 상황에서 HUMIRA 치료 재개를 고려할 때 주의하고 환자를 면밀히 모니터링하십시오.
5.5 신경계 반응
HUMIRA를 포함한 TNF 차단제의 사용은 다발성 경화증(MS) 및 시신경염을 포함한 중추 신경계 탈수초성 질환과 길랑-바레 증후군을 포함한 말초 탈수초성 질환의 새로운 발병 또는 임상 증상 악화 및/또는 방사선학적 증거의 드문 사례와 관련이 있습니다. 기존 또는 최근 발병한 중추 또는 말초 신경계 탈수초성 질환이 있는 환자에게 HUMIRA 사용을 고려할 때 주의하십시오. 이러한 질환 중 하나라도 발생하면 HUMIRA를 중단하는 것을 고려해야 합니다. 중간 포도막염과 중추 탈수초성 질환 사이에는 알려진 연관성이 있습니다.
5.6 혈액학적 반응
재생불량성 빈혈을 포함한 범혈구감소증의 드문 사례가 TNF 차단제에서 보고되었습니다. 의학적으로 유의미한 세포감소증(예: 혈소판 감소증, 백혈구 감소증)을 포함한 혈액계의 이상 반응이 HUMIRA에서 드물게 보고되었습니다. 이러한 보고와 HUMIRA의 인과 관계는 불분명합니다. HUMIRA를 복용하는 동안 혈액 질환이나 감염(예: 지속적인 발열, 멍, 출혈, 창백함)을 암시하는 징후와 증상이 나타나면 즉시 의사의 진료를 받도록 모든 환자에게 조언하십시오. 유의미한 혈액학적 이상이 확인된 환자의 경우 HUMIRA 치료 중단을 고려하십시오.
5.7 Anakinra와 함께 사용할 경우 감염 위험 증가
인터루킨-1 길항제인 anakinra와 다른 TNF 차단제의 병용 사용은 RA 환자에서 TNF 차단제 단독에 비해 심각한 감염 및 호중구 감소증의 비율이 더 높았고 추가적인 이점은 없었습니다. 따라서 HUMIRA와 anakinra의 병용은 권장되지 않습니다 [see Drug Interactions (7.2)].
5.8 심부전
울혈성 심부전(CHF) 악화 및 새로 발생한 CHF 사례가 TNF 차단제에서 보고되었습니다. CHF 악화 사례는 HUMIRA에서도 관찰되었습니다. HUMIRA는 CHF 환자를 대상으로 공식적으로 연구된 적이 없습니다. 그러나 다른 TNF 차단제의 임상 시험에서 심각한 CHF 관련 이상 반응의 비율이 더 높게 관찰되었습니다. 심부전이 있는 환자에게 HUMIRA를 사용할 때는 주의하고 주의 깊게 모니터링하십시오.
5.9 자가면역
HUMIRA로 치료하면 자가항체가 형성될 수 있으며 드물게 루푸스 유사 증후군이 발생할 수 있습니다. HUMIRA로 치료한 후 환자에게 루푸스 유사 증후군을 시사하는 증상이 나타나면 치료를 중단하십시오 [see Adverse Reactions (6.1)].
5.10 예방 접종
RA 환자를 대상으로 한 위약 대조 임상 시험에서 폐렴구균 다당체 백신과 인플루엔자 백신을 HUMIRA와 동시에 투여했을 때 HUMIRA 치료군과 위약 치료군 간에 항폐렴구균 항체 반응의 차이가 나타나지 않았습니다. HUMIRA 치료군과 위약 치료군 간에 비슷한 비율의 환자가 인플루엔자 항체에 대한 방어 수준을 개발했습니다. 그러나 HUMIRA를 투여받은 환자에서 인플루엔자 항원에 대한 전체 역가는 약간 낮았습니다. 이것의 임상적 중요성은 알려져 있지 않습니다. HUMIRA를 투여받는 환자는 생백신을 제외하고 동시에 예방 접종을 받을 수 있습니다. HUMIRA를 투여받는 환자에서 생백신에 의한 감염의 2차 전파에 대한 데이터는 없습니다.
소아 환자의 경우 가능하면 HUMIRA 치료를 시작하기 전에 현재 예방 접종 지침에 따라 모든 예방 접종을 최신 상태로 유지하는 것이 좋습니다. HUMIRA를 투여받는 환자는 생백신을 제외하고 동시에 예방 접종을 받을 수 있습니다.
자궁 내에서 HUMIRA에 노출된 영아에게 생백신 또는 약독화 생백신을 투여하는 것의 안전성은 알려져 있지 않습니다. 노출된 영아에게 백신(생백신 또는 약독화 생백신)을 접종하기 전에 위험과 이점을 고려해야 합니다 [see Use in Specific Populations (8.1, 8.4)].
5.11 아바타셉트와 병용 시 감염 위험 증가
대조 임상시험에서 TNF 차단제와 아바타셉트의 병용 투여는 TNF 차단제 단독 사용보다 심각한 감염의 비율이 더 높았습니다. 병용 요법은 TNF 차단제 단독 사용에 비해 RA 치료에서 임상적 이점을 보여주지 못했습니다. 따라서 HUMIRA를 포함한 TNF 차단제와 아바타셉트의 병용은 권장되지 않습니다 [7.2] 약물 상호 작용 참조.
6. 부작용
다음 임상적으로 중요한 부작용은 라벨링의 다른 부분에서 설명됩니다.
- 중증 감염 [경고 및 주의 사항 (5.1)]
- 악성 종양 [경고 및 주의 사항 (5.2)]
- 과민 반응 [경고 및 주의 사항 (5.3)]
- B형 간염 바이러스 재활성화 [경고 및 주의 사항 (5.4)]
- 신경계 반응 [경고 및 주의 사항 (5.5)]
- 혈액학적 반응 [경고 및 주의 사항 (5.6)]
- 심부전 [경고 및 주의 사항 (5.8)]
- 자가 면역 [경고 및 주의 사항 (5.9)]
6.1 임상 시험 경험
임상 시험은 매우 다양한 조건에서 수행되므로, 약물의 임상 시험에서 관찰된 부작용 발생률은 다른 약물의 임상 시험에서 관찰된 발생률과 직접 비교할 수 없으며 실제로 관찰된 발생률을 반영하지 않을 수 있습니다.
HUMIRA의 가장 흔한 부작용은 주사 부위 반응이었습니다. 위약 대조 임상 시험에서 HUMIRA로 치료받은 환자의 20%에서 주사 부위 반응(발적 및/또는 가려움증, 출혈, 통증 또는 부종)이 발생한 반면, 위약을 투여받은 환자의 14%에서 발생했습니다. 대부분의 주사 부위 반응은 경미한 것으로 묘사되었으며 일반적으로 약물 중단을 요구하지 않았습니다.
RA 환자(즉, 연구 RA-I, RA-II, RA-III 및 RA-IV)에서 이루어진 이중맹검, 위약 대조 연구의 이중맹검, 위약 대조 부분 동안 부작용으로 인해 치료를 중단한 환자의 비율은 HUMIRA를 복용한 환자의 경우 7%, 위약을 투여받은 환자의 경우 4%였습니다. 이러한 RA 연구에서 HUMIRA 중단으로 이어진 가장 흔한 부작용은 임상적 악화 반응(0.7%), 발진(0.3%) 및 폐렴(0.3%)이었습니다.
감염
RA, PsA, AS, CD, UC, Ps, HS 및 UV 성인 환자를 대상으로 한 39건의 글로벌 HUMIRA 임상 시험의 대조군 부분에서, 중증 감염 발생률은 HUMIRA 치료를 받은 7973명의 환자에서 100 환자-년당 4.3건이었고, 대조군 치료를 받은 4848명의 환자에서 100 환자-년당 2.9건이었습니다. 관찰된 중증 감염에는 폐렴, 패혈성 관절염, 보철 및 수술 후 감염, 단독, 봉와직염, 게실염 및 신우신염이 포함되었습니다. [경고 및 주의 사항 (5.1)].
결핵 및 기회 감염
RA, PsA, AS, CD, UC, Ps, HS 및 UV를 포함한 24,605명의 HUMIRA 치료를 받은 환자를 대상으로 한 52건의 글로벌 대조군 및 비대조군 임상 시험에서, 보고된 활동성 결핵 발생률은 100 환자-년당 0.20건이었고, 양성 PPD 전환 발생률은 100 환자-년당 0.09건이었습니다. 10,113명의 미국 및 캐나다 HUMIRA 치료를 받은 환자의 하위 그룹에서, 보고된 활동성 결핵 발생률은 100 환자-년당 0.05건이었고, 양성 PPD 전환 발생률은 100 환자-년당 0.07건이었습니다. 이러한 시험에는 폐결핵, 림프절 결핵, 복막 결핵 및 폐결핵 보고가 포함되었습니다. 대부분의 결핵 사례는 치료 시작 후 8개월 이내에 발생했으며 잠복 결핵의 재발을 반영할 수 있습니다. 이러한 글로벌 임상 시험에서, 중증 기회 감염 사례가 100 환자-년당 0.05건의 전반적인 발생률로 보고되었습니다. 중증 기회 감염 및 결핵 사례 중 일부는 치명적이었습니다. [경고 및 주의 사항 (5.1)].
자가 항체
류마티스 관절염 대조군 시험에서, 기준선 ANA 역가가 음성인 HUMIRA 치료를 받은 환자의 12%와 위약 치료를 받은 환자의 7%에서 24주째에 양성 역가가 나타났습니다. HUMIRA로 치료받은 3046명 중 2명의 환자에서 새로 발생한 루푸스 유사 증후군을 시사하는 임상적 징후가 나타났습니다. 환자는 치료 중단 후 호전되었습니다. 루푸스 신염이나 중추 신경계 증상이 발생한 환자는 없었습니다. HUMIRA로 장기간 치료한 경우 자가 면역 질환 발생에 미치는 영향은 알려져 있지 않습니다.
간 효소 상승
TNF 억제제를 투여받은 환자에서 급성 간부전을 포함한 심각한 간 반응이 보고되었습니다. RA, PsA 및 AS 환자를 대상으로 한 HUMIRA(40mg SC 격주)의 대조군 3상 시험에서, 대조군 기간 지속 시간이 4주에서 104주까지인 경우, HUMIRA 치료를 받은 환자의 3.5%와 대조군 치료를 받은 환자의 1.5%에서 ALT 상승 ≥ 3 x ULN이 발생했습니다. 이러한 시험에서 많은 환자가 간 효소 상승을 유발하는 약물(예: NSAID, MTX)을 복용하고 있었기 때문에, HUMIRA와 간 효소 상승 간의 관계는 명확하지 않습니다. 4세에서 17세의 다관절 JIA 환자를 대상으로 한 HUMIRA의 대조군 3상 시험에서, HUMIRA 치료를 받은 환자의 4.4%와 대조군 치료를 받은 환자의 1.5%에서 ALT 상승 ≥ 3 x ULN이 발생했습니다(ALT가 AST보다 흔함). 간 효소 검사 상승은 HUMIRA와 MTX를 병용 투여한 환자에서 HUMIRA 단독 투여한 환자보다 더 빈번했습니다. 일반적으로 이러한 상승은 HUMIRA 치료 중단으로 이어지지 않았습니다. 2세에서 <4세의 다관절 JIA 환자를 대상으로 한 HUMIRA의 공개 라벨 연구에서는 ALT 상승 ≥ 3 x ULN이 발생하지 않았습니다.
HUMIRA (160 mg 및 80 mg의 초기 용량 또는 1일 및 15일에 각각 80 mg 및 40 mg, 그 후 격주로 40 mg)의 대조군 기간 지속 시간이 4주에서 52주까지인 크론병 성인 환자를 대상으로 한 제3상 대조군 시험에서 ALT 상승 ≥ 3 x ULN은 HUMIRA 치료 환자의 0.9%와 대조군 치료 환자의 0.9%에서 발생했습니다. 최대 52주까지 치료를 받은 체중 기반 유도 요법 후 체중 기반 유지 용량 요법 2가지에 대한 효능 및 안전성을 평가한 크론병 소아 환자를 대상으로 한 HUMIRA의 제3상 시험에서 ALT 상승 ≥ 3 x ULN은 환자의 2.6%(5/192)에서 발생했으며, 이 중 4명은 기준선에서 면역 억제제를 병용했습니다. 이러한 환자 중 ALT 검사 이상으로 인해 치료를 중단한 환자는 없었습니다. HUMIRA (1일 및 15일에 각각 160 mg 및 80 mg의 초기 용량, 그 후 격주로 40 mg)의 대조군 기간 지속 시간이 1주에서 52주까지인 궤양성 대장염 성인 환자를 대상으로 한 제3상 대조군 시험에서 ALT 상승 ≥ 3 x ULN은 HUMIRA 치료 환자의 1.5%와 대조군 치료 환자의 1.0%에서 발생했습니다. 격주로 0.6 mg/kg(최대 40 mg)의 유지 용량(N=31)과 매주 0.6 mg/kg(최대 40 mg)의 유지 용량(N=32)에 대한 효능 및 안전성을 평가한 소아 궤양성 대장염 환자(N=93)를 대상으로 한 제3상 대조군 시험에서 0주와 1주에 2.4 mg/kg(최대 160 mg)의 체중 기반 유도 용량과 2주에 1.2 mg/kg(최대 80 mg)의 체중 기반 유도 용량(N=63) 또는 0주에 2.4 mg/kg(최대 160 mg)의 유도 용량, 1주에 위약, 2주에 1.2 mg/kg(최대 80 mg)의 유도 용량(N=30)을 투여한 후 ALT 상승 ≥ 3 X ULN은 환자의 1.1%(1/93)에서 발생했습니다. HUMIRA (80 mg의 초기 용량, 그 후 격주로 40 mg)의 대조군 기간 지속 시간이 12주에서 24주까지인 건선 환자를 대상으로 한 제3상 대조군 시험에서 ALT 상승 ≥ 3 x ULN은 HUMIRA 치료 환자의 1.8%와 대조군 치료 환자의 1.8%에서 발생했습니다. 대조군 기간 지속 시간이 12주에서 16주까지인 hidradenitis suppurativa 환자를 대상으로 한 HUMIRA (0주에 160 mg, 2주에 80 mg의 초기 용량, 그 후 4주부터 매주 40 mg)의 대조군 시험에서 ALT 상승 ≥ 3 x ULN은 HUMIRA 치료 환자의 0.3%와 대조군 치료 환자의 0.6%에서 발생했습니다. HUMIRA 치료 환자와 대조군 치료 환자의 노출 기간이 각각 165.4 PY와 119.8 PY인 포도막염 성인 환자를 대상으로 한 HUMIRA (0주에 80 mg의 초기 용량, 그 후 1주부터 격주로 40 mg)의 대조군 시험에서 ALT 상승 ≥ 3 x ULN은 HUMIRA 치료 환자의 2.4%와 대조군 치료 환자의 2.4%에서 발생했습니다.
기타 이상 반응
류마티스 관절염 임상 연구
아래에 설명된 데이터는 2468명의 환자(6개월 동안 노출된 환자 2073명, 1년 이상 노출된 환자 1497명, 적절하고 잘 통제된 연구(연구 RA-I, RA-II, RA-III 및 RA-IV)에 참여한 환자 1380명 포함)에서 HUMIRA에 대한 노출을 반영합니다. HUMIRA는 주로 위약 대조군 시험과 최대 36개월 지속 기간의 장기 추적 연구에서 연구되었습니다. 모집단의 평균 연령은 54세였으며, 여성이 77%, 백인이 91%였으며, 중등도에서 중증의 활동성 류마티스 관절염을 앓고 있었습니다. 대부분의 환자는 격주로 40 mg HUMIRA를 투여받았습니다 [임상 연구 ( 14.1)] 참조].
표 1은 위약과 비교하여 격주로 40 mg HUMIRA를 투여받은 환자에서 발생률이 5% 이상이고 위약보다 발생률이 높은 반응을 요약합니다. 연구 RA-III에서 2년차 오픈 라벨 연장 연구의 이상 반응 유형 및 빈도는 1년차 이중 맹검 부분에서 관찰된 것과 유사했습니다.
| HUMIRA 40 mg 피하 격주 투여 |
위약 | |
| (N=705) | (N=690) | |
| 이상 반응(선호 용어) | ||
| 호흡기 | ||
| 상기도 감염 | 17% | 13% |
| 축농증 | 11% | 9% |
| 독감 증후군 | 7% | 6% |
| 위장 | ||
| 메스꺼움 | 9% | 8% |
| 복통 | 7% | 4% |
| 검사실 검사* | ||
| 검사실 검사 이상 | 8% | 7% |
| 고콜레스테롤혈증 | 6% | 4% |
| 고지혈증 | 7% | 5% |
| 혈뇨 | 5% | 4% |
| 알칼리성 인산 가수분해효소 증가 | 5% | 3% |
| 기타 | ||
| 두통 | 12% | 8% |
| 발진 | 12% | 6% |
| 우발적 부상 | 10% | 8% |
| 주사 부위 반응 ** | 8% | 1% |
| 요통 | 6% | 4% |
| 요로 감염 | 8% | 5% |
| 고혈압 | 5% | 3% |
| * 유럽 임상 시험에서 부작용으로 보고된 실험실 검사 이상 ** 주사 부위 홍반, 가려움증, 출혈, 통증 또는 붓기를 포함하지 않음 |
||
류마티스 관절염 임상 연구에서 덜 흔한 유해 반응
RA 연구에서 HUMIRA 치료를 받은 환자에서 5% 미만의 발생률로 나타났지만 경고 및 주의 사항 또는 유해 반응 섹션에 나타나지 않은 다른 드문 심각한 유해 반응은 다음과 같습니다.
전신: 사지 통증, 골반 통증, 수술, 흉곽 통증
심혈관계: 부정맥, 심방 세동, 흉통, 관상 동맥 질환, 심장 마비, 고혈압성 뇌병증, 심근 경색, 두근거림, 심낭 삼출, 심낭염, 실신, 빈맥
소화기계: 담낭염, 담석증, 식도염, 위장염, 위장관 출혈, 간 괴사, 구토
내분비계: 부갑상선 장애
혈액 및 림프계: 무과립구증, 다혈구증
대사 및 영양 장애: 탈수, 치유 이상, 케톤증, 단백질혈증, 말초 부종
근골격계: 관절염, 골 장애, 골절 (자발적이지 않음), 골 괴사, 관절 장애, 근육 경련, 근무력증, 화농성 관절염, 활막염, 힘줄 장애
신생물: 선종
신경계: 혼돈, 감각 이상, 경막하 출혈, 떨림
호흡기계: 천식, 기관지 경련, 호흡 곤란, 폐 기능 감소, 흉막 삼출
특수 감각: 백내장
혈전증: 다리 혈전증
비뇨생식기계: 방광염, 신장 결석, 월경 장애
소아 특발성 관절염 임상 연구
일반적으로 다관절 소아 특발성 관절염 (JIA) 시험 (JIA-I 및 JIA-II 연구) [임상 연구 참조 (14.2)]에서 HUMIRA 치료를 받은 환자의 유해 반응은 성인 환자에서 관찰된 빈도 및 유형과 유사했습니다. [경고 및 주의 사항 참조 (5), 유해 반응 (6)]. 성인과의 중요한 발견 사항과 차이점은 다음 단락에서 논의됩니다.
JIA-I 연구에서 HUMIRA는 다관절 JIA를 가진 4세에서 17세 사이의 171명의 환자에서 연구되었습니다. 연구에서 보고된 심각한 유해 반응에는 호중구 감소증, 연쇄상 구균 인두염, 아미노 전이효소 증가, 대상 포진, 근육염, 자궁 출혈 및 맹장염이 포함되었습니다. 심각한 감염은 HUMIRA 치료 시작 후 약 2년 이내에 환자의 4%에서 관찰되었으며 단순 포진, 폐렴, 요로 감염, 인두염 및 대상 포진 사례가 포함되었습니다.
JIA-I 연구에서 환자의 45%가 HUMIRA 치료를 받는 동안 첫 16주 동안 MTX를 동시에 사용하거나 사용하지 않고 감염을 경험했습니다. HUMIRA 치료를 받은 환자에서 보고된 감염 유형은 일반적으로 TNF 억제제로 치료받지 않은 다관절 JIA 환자에서 흔히 볼 수 있는 감염과 유사했습니다. 치료 시작 시 HUMIRA로 치료받은 이 환자군에서 가장 흔하게 발생하는 유해 반응은 주사 부위 통증과 주사 부위 반응 (각각 19% 및 16%)이었습니다. HUMIRA를 투여받은 환자에서 덜 흔하게 보고된 유해 사건은 육아종성 환상이었으며 HUMIRA 치료 중단으로 이어지지 않았습니다.
JIA-I 연구에서 치료 첫 48주 동안 비 심각한 과민 반응이 환자의 약 6%에서 관찰되었으며 주로 국소 알레르기 과민 반응 및 알레르기 발진이 포함되었습니다.
JIA-I 연구에서 기준선 항-dsDNA 항체가 음성인 HUMIRA 치료를 받은 환자의 10%가 치료 48주 후 양성 역가를 나타냈습니다. 임상 시험 동안 어떤 환자도 자가 면역 질환의 임상 증상을 보이지 않았습니다.
JIA-I 연구에서 HUMIRA 치료를 받은 환자의 약 15%가 경증에서 중등도의 크레아틴 키나아제 (CPK) 상승을 보였습니다. 여러 환자에서 정상 상한치의 5배를 초과하는 상승이 관찰되었습니다. 모든 환자에서 CPK 농도가 감소하거나 정상으로 돌아왔습니다. 대부분의 환자는 HUMIRA 치료를 중단하지 않고 계속할 수 있었습니다.
JIA-II 연구에서 HUMIRA는 다관절 JIA를 가진 2세에서 <4세 또는 4세 이상으로 체중이 <15kg인 32명의 환자에서 연구되었습니다. 이 환자군의 안전성 프로파일은 다관절 JIA를 가진 4세에서 17세 사이의 환자에서 관찰된 안전성 프로파일과 유사했습니다.
JIA-II 연구에서 환자의 78%가 HUMIRA 치료를 받는 동안 감염을 경험했습니다. 여기에는 비인두염, 기관지염, 상기도 감염, 중이염이 포함되었으며 대부분 경증에서 중등도였습니다. 연구에서 HUMIRA를 투여받은 환자의 9%에서 심각한 감염이 관찰되었으며 치아 우식증, 로타 바이러스 위장염 및 수두가 포함되었습니다.
JIA-II 연구에서 비 심각한 알레르기 반응이 환자의 6%에서 관찰되었으며 간헐적 두드러기 및 발진이 포함되었으며 모두 경증이었습니다.
건선성 관절염 및 강직성 척추염 임상 연구
HUMIRA는 두 건의 위약 대조 시험과 개방 표지 연구에서 건선성 관절염 (PsA) 환자 395명과 두 건의 위약 대조 시험에서 강직성 척추염 (AS) 환자 393명에서 연구되었습니다. [임상 연구 참조 (14.3, 14.4)]. HUMIRA 40mg을 격주로 투여받은 PsA 및 AS 환자의 안전성 프로파일은 RA 환자에서 관찰된 안전성 프로파일과 유사했습니다. HUMIRA 연구 RA-I에서 IV까지.
크론병 임상 연구
성인: 네 건의 위약 대조 시험과 두 건의 개방 표지 연장 연구에서 크론병 환자 1478명의 HUMIRA 안전성 프로파일 [임상 연구 참조 (14.5)] 은 RA 환자에서 관찰된 안전성 프로파일과 유사했습니다.
6세에서 17세까지의 소아 환자: HUMIRA의 안전성 프로파일은 한 건의 이중맹검 연구(연구 PCD-I)와 한 건의 오픈 라벨 연장 연구에서 192명의 소아 환자에서 [임상 연구 (14.6)] 크론병 성인 환자에서 관찰된 안전성 프로파일과 유사했습니다.
연구 PCD-I의 4주 오픈 라벨 유도 단계에서 HUMIRA로 치료받은 소아 인구에서 가장 흔하게 발생한 부작용은 주사 부위 통증과 주사 부위 반응(각각 6%와 5%)이었습니다.
연구 PCD-I에서 HUMIRA를 투여받는 동안 총 67%의 어린이가 감염을 경험했습니다. 여기에는 상기도 감염과 비인두염이 포함됩니다.
연구 PCD-I에서 HUMIRA를 투여받는 동안 총 5%의 어린이가 심각한 감염을 경험했습니다. 여기에는 바이러스 감염, 기기 관련 패혈증(카테터), 위장염, H1N1 인플루엔자 및 파종성 히스토플라스마증이 포함됩니다.
연구 PCD-I에서 알레르기 반응은 어린이의 5%에서 관찰되었으며, 모두 심각하지 않았으며 주로 국소 반응이었습니다.
궤양성 대장염 임상 연구
성인: HUMIRA의 안전성 프로파일은 두 건의 위약 대조 연구와 한 건의 오픈 라벨 연장 연구에서 궤양성 대장염(UC)이 있는 1010명의 성인 환자에서 [임상 연구 (14.7)] RA 환자에서 관찰된 안전성 프로파일과 유사했습니다.
5세에서 17세까지의 소아 환자: HUMIRA의 안전성 프로파일은 한 건의 이중맹검 연구와 한 건의 오픈 라벨 연장 연구에서 궤양성 대장염이 있는 93명의 소아 환자에서 [임상 연구 (14.8)] 궤양성 대장염 성인 환자에서 관찰된 안전성 프로파일과 유사했습니다.
판상 건선 임상 연구
HUMIRA는 위약 대조 및 오픈 라벨 연장 연구에서 판상 건선(Ps)이 있는 1696명의 피험자에서 연구되었습니다. [임상 연구 (14.9)]. Ps로 치료받은 피험자의 HUMIRA에 대한 안전성 프로파일은 RA 피험자에서 관찰된 안전성 프로파일과 유사했으며, 다음과 같은 예외가 있었습니다. Ps 피험자에 대한 임상 시험의 위약 대조 부분에서 HUMIRA로 치료받은 피험자는 대조군에 비해 관절통의 발생률이 더 높았습니다(3% vs. 1%).
농포성 한선염 임상 연구
HUMIRA는 세 건의 위약 대조 연구와 한 건의 오픈 라벨 연장 연구에서 농포성 한선염(HS)이 있는 727명의 피험자에서 연구되었습니다. [임상 연구 (14.10)]. HUMIRA로 매주 치료받은 HS 피험자의 안전성 프로파일은 HUMIRA의 알려진 안전성 프로파일과 일치했습니다.
HS의 악화는 기준선에서 농양 및 염증성 결절 수의 ≥25% 증가 및 최소 2개의 추가 병변이 있는 것으로 정의되었으며, 두 연구에서 1차 유효성 평가 시점 이후 HUMIRA 치료를 중단한 100명의 피험자 중 22명(22%)에서 문서화되었습니다.
포도막염 임상 연구
HUMIRA는 위약 대조 및 오픈 라벨 연장 연구에서 포도막염(UV)이 있는 464명의 성인 환자와 포도막염(연구 PUV-I)이 있는 90명의 소아 환자에서 연구되었습니다. [임상 연구 (14.11, 14.12)] . HUMIRA로 치료받은 UV 환자의 안전성 프로파일은 RA 환자에서 관찰된 안전성 프로파일과 유사했습니다.
6.2 면역원성
모든 치료용 단백질과 마찬가지로 면역원성이 발생할 가능성이 있습니다. 항체 형성의 검출은 검사의 민감도와 특이도에 크게 의존합니다. 또한, 검사에서 관찰된 항체(중화 항체 포함) 양성률은 검사 방법, 샘플 취급, 샘플 수집 시기, 동반 약물 및 기저 질환을 포함한 여러 요인의 영향을 받을 수 있습니다. 이러한 이유로 아래에 설명된 연구에서 항체 발생률을 다른 연구의 항체 발생률 또는 다른 아달리무맙 제품과 비교하는 것은 오해의 소지가 있을 수 있습니다.
아달리무맙에 대한 항체를 측정하는 데 사용된 두 가지 검사가 있습니다. ELISA를 사용하면 혈청 아달리무맙 농도가 < 2 mcg/mL일 때만 아달리무맙에 대한 항체를 검출할 수 있었습니다. ECL 검사는 혈청 샘플에서 아달리무맙 농도에 관계없이 아달리무맙에 대한 항체 역가를 검출할 수 있습니다. HUMIRA로 치료받은 환자에서 아달리무맙에 대한 항체(AAA) 발생률은 표 2에 제시되어 있습니다.
| 적응증 | 연구 기간 | ELISA에 의한 아달리무맙에 대한 항체 발생률 (n/N) | ECL 검사에 의한 아달리무맙에 대한 항체 발생률 (n/N) | |||||
| 아달리무맙을 투여받은 모든 환자 | 혈청 아달리무맙 농도가 < 2 mcg/mL인 환자 | |||||||
| 류마티스 관절염a |
6개월에서 12개월 | 5% (58/1062) | NR | NA | ||||
| 소아 특발성 관절염 (JIA) | 4세에서 17세b | 48주 | 16% (27/171) | NR | NA |
| 2세에서 4세 또는 ≥ 4세이고 체중이 < 15kg | 24주 | 7% (1/15)c | NR | NA | |
| 건선성 관절염d | 48주e | 13% (24/178) | NR | NA | |
| 강직성 척추염 | 24주 | 9% (16/185) | NR | NA | |
| 성인 크론병 | 56주 | 3% (7/269) | 8% (7/86) | NA | |
| 소아 크론병 | 52주 | 3% (6/182) | 10% (6/58) | NA | |
| 성인 궤양성 대장염 | 52주 | 5% (19/360) | 21% (19/92) | NA | |
| 소아 궤양성 대장염 | 52주 | 3% (3/100) | 13% (3/23) | 33% (33/100)i | |
| 판상 건선f | 최대 52주g | 8% (77/920) | 21% (77/372) | NA | |
| 농포성 한선염 | 36주 | 7% (30/461) | 28% (58/207)h | 61% (272/445)j | |
| 비감염성 포도막염 | 52주 | 5% (12/249) | 21% (12/57) | 40% (99/249)k | |
n: 항-아달리무맙 항체를 가진 환자 수; NR: 보고되지 않음; NA: 해당 없음(실시하지 않음)
a 메토트렉세이트(MTX)를 병용 투여받은 환자의 경우, 항-아달리무맙 항체 발생률은 HUMIRA 단독 요법의 경우 12%에 비해 1%였다.
b 메토트렉세이트(MTX)를 병용 투여받은 환자의 경우, 항-아달리무맙 항체 발생률은 HUMIRA 단독 요법의 경우 26%에 비해 6%였다.
c 이 환자는 메토트렉세이트(MTX)를 병용 투여받았다.
d 메토트렉세이트(MTX)를 병용 투여받은 환자의 경우, 항체 발생률은 RA의 경우 1%에 비해 7%였다.
e 24주 또는 12주 치료의 이전 연구 2건을 완료한 후 등록된 피험자.
f HUMIRA 단독 요법을 받았던 플라크 건선 환자에서 치료를 중단한 후 재치료 시 아달리무맙에 대한 항체 발생률은 중단 전에 관찰된 발생률과 유사했다.
g 12주 2상 연구 1건과 52주 3상 연구 1건
h 2건의 3상 연구에서 최대 24주 동안 HUMIRA 치료를 중단하고 아달리무맙 혈청 수치가 이후 <2 mcg/mL(연구 대상 전체 피험자의 약 22%)로 감소한 피험자 중
i 항체 발생과 안전성 사이에 명확한 연관성은 관찰되지 않았다. 항체 발생과 효능 결과의 연관성은 항-아달리무맙 항체 역가에 따라 계층화된 각 치료군의 피험자 수가 제한적이어서 평가하지 않았다.
j 항체 발생과 안전성 사이에 명확한 연관성은 관찰되지 않았다.
k 항체 발생과 안전성 또는 효능 결과 사이의 상관관계는 관찰되지 않았다.
류마티스 관절염 및 건선성 관절염: 연구 RA-I, RA-II 및 RA-III의 환자는 6~12개월 동안 ELISA를 사용하여 아달리무맙에 대한 항체를 여러 시점에서 검사했다. 항체 발생과 이상 반응 사이의 명확한 상관관계는 관찰되지 않았다. 단독 요법의 경우, 매주 투여를 받는 환자보다 격주 투여를 받는 환자에서 항체가 더 자주 발생할 수 있다. 단독 요법으로 권장 용량인 40mg을 격주로 투여받은 환자의 경우, 항체 양성 환자보다 항체 음성 환자에서 ACR 20 반응이 더 낮았다. HUMIRA의 장기 면역원성은 알려져 있지 않다.
6.3 시판 후 경험
HUMIRA의 시판 후 사용 중 다음과 같은 이상 반응이 확인되었다. 이러한 반응은 불확실한 규모의 모집단에서 자발적으로 보고되기 때문에, 항상 발생 빈도를 신뢰할 수 있게 추정하거나 HUMIRA 노출과의 인과 관계를 확립할 수 있는 것은 아니다.
위장관 장애: 맹장염, 대장 천공(맹장염과 관련된 천공 및 충수염과 관련된 충수 천공 포함), 췌장염
일반 장애 및 투여 부위 상태: 발열
간담도계 장애: 간부전, 간염
면역 체계 장애: 사르코이드증
신생물(양성, 악성 및 불명확)(낭포 및 용종 포함): 머클 세포 암종(피부의 신경내분비 암종)
신경계 장애: 탈수초 질환(예: 시신경염, 길랭-바레 증후군), 뇌혈관 사고
호흡기 장애: 간질성 폐 질환(폐 섬유증 포함), 폐색전증
피부 반응: 스티븐스-존슨 증후군, 피부 혈관염, 다형 홍반, 새로운 또는 악화된 건선(농포성 및 손바닥-발바닥형 포함), 탈모, 지루성 피부 반응
혈관 장애: 전신 혈관염, 심부 정맥 혈전증
7. 약물 상호 작용
7.1 메토트렉세이트
HUMIRA는 메토트렉세이트(MTX)를 병용 투여하는 류마티스 관절염(RA) 환자에서 연구되었습니다. MTX는 아달리무맙의 명백한 클리어런스를 감소시켰지만, 데이터는 HUMIRA 또는 MTX의 용량 조정이 필요하지 않음을 시사하지 않습니다 [임상 약리학 (12.3)].
7.2 생물학적 제제
RA 환자를 대상으로 한 임상 연구에서 TNF 억제제와 아나킨라 또는 아바타셉트를 병용 투여하면 심각한 감염 위험이 증가하는 것으로 나타났으며, 추가적인 이점은 없었습니다. 따라서 RA 환자에서 HUMIRA와 아바타셉트 또는 아나킨라를 병용하는 것은 권장되지 않습니다 [경고 및 주의 사항 (5.7, 5.11)]. 또한 리툭시맙으로 치료받은 RA 환자에서 TNF 억제제로 후속 치료를 받으면 심각한 감염 발생률이 높아지는 것으로 나타났습니다. RA, PsA, AS, CD, UC, Ps, HS 및 UV 치료를 위한 HUMIRA와 다른 생물학적 제제의 병용 사용에 대한 정보는 충분하지 않습니다. 감염 위험 증가 및 기타 잠재적인 약리학적 상호 작용 가능성을 고려하여 HUMIRA와 다른 생물학적 DMARDS(예: 아나킨라 및 아바타셉트) 또는 다른 TNF 억제제를 병용 투여하는 것은 권장되지 않습니다.
7.4 시토크롬 P450 기질
만성 염증 동안 사이토카인(예: TNFα, IL-6)의 농도가 증가하면 CYP450 효소의 형성이 억제될 수 있습니다. 아달리무맙과 같이 사이토카인 활성을 길항하는 분자가 CYP450 효소의 형성에 영향을 미칠 수 있습니다. CYP450 기질(좁은 치료 범위)로 치료받는 환자에서 HUMIRA를 시작하거나 중단할 때, 효과(예: 와파린) 또는 약물 농도(예: 사이클로스포린 또는 테오필린)를 모니터링하는 것이 권장되며, 필요에 따라 약물 제품의 개별 용량을 조정할 수 있습니다.
8 특정 인구 집단에서의 사용
8.1 임신
위험 요약
임신 중 아달리무맙 사용에 대한 이용 가능한 연구는 아달리무맙과 주요 선천적 기형 사이의 연관성을 확실하게 밝히지 못합니다. 임상 데이터는 류마티스 관절염(RA) 또는 크론병(CD)이 있는 임산부에 대한 Organization of Teratology Information Specialists (OTIS)/MotherToBaby HUMIRA 임신 레지스트리에서 얻을 수 있습니다. 레지스트리 결과는 RA 또는 CD가 있는 임산부에서 아달리무맙을 1분기 동안 사용한 경우 주요 선천적 기형 발생률이 10%이고, 질병 일치 비교 코호트에서 주요 선천적 기형 발생률이 7.5%임을 보여주었습니다. 주요 선천적 기형의 패턴이 없다는 것은 안심할 만하며, 노출 그룹 간의 차이가 선천적 기형 발생에 영향을 미쳤을 수 있습니다 (자료 참조).
아달리무맙은 임신 3분기 동안 태반을 통해 활발하게 이동하며, 자궁 내 노출된 영아의 면역 반응에 영향을 미칠 수 있습니다 (임상적 고려 사항 참조). 시험관 원숭이에서 수행된 배아-태아 출산 전후 발달 연구에서, 기관 형성 과정과 임신 후기에 메토트렉세이트 없이 피하 주사로 최대 권장 인간 용량(MRHD) 40mg의 약 373배에 해당하는 노출을 유발하는 용량으로 아달리무맙을 정맥 주사했을 때 태아에게 해를 입히거나 기형이 발생하지 않았습니다 (자료 참조).
지정된 모집단에 대한 주요 선천적 기형 및 유산의 추정 배경 위험은 알려져 있지 않습니다. 모든 임신에는 선천적 기형, 손실 또는 기타 부작용의 배경 위험이 있습니다. 미국 일반 모집단에서 임상적으로 인식된 임신에서 주요 선천적 기형 및 유산의 추정 배경 위험은 각각 2-4% 및 15-20%입니다.
임상적 고려 사항
질병 관련 모체 및 배아/태아 위험
발표된 데이터에 따르면 RA 또는 염증성 장 질환(IBD)이 있는 여성에서의 불리한 임신 결과 위험은 질병 활동 증가와 관련이 있습니다. 불리한 임신 결과에는 조산(임신 37주 전), 저체중아(2500g 미만) 출산 및 출생 시 태아 성장 지연이 포함됩니다.
태아/신생아 부작용
단일클론 항체는 임신이 진행됨에 따라 태반을 통해 점점 더 많이 이동하며, 3분기에 가장 많은 양이 이동합니다 (자료 참조). HUMIRA에 자궁 내 노출된 영아에게 생백신 또는 생독소 백신을 투여하기 전에 위험과 이점을 고려해야 합니다 [특정 모집단에서의 사용 (8.4)].
자료
인간 데이터
2004년부터 2016년까지 미국과 캐나다에서 OTIS/MotherToBaby가 수행한 전향적 코호트 임신 노출 레지스트리는 1분기에 아달리무맙으로 치료받은 여성 221명(RA 69명, CD 152명)과 아달리무맙으로 치료받지 않은 여성 106명(RA 74명, CD 32명)의 생존 영아에서 주요 선천적 기형 위험을 비교했습니다.
아달리무맙으로 치료받은 코호트와 치료받지 않은 코호트에서 생존 영아 중 주요 선천적 기형 비율은 각각 10%(RA 8.7%, CD 10.5%)와 7.5%(RA 6.8%, CD 9.4%)였습니다. 주요 선천적 기형의 패턴이 없다는 것은 안심할 만하며, 노출 그룹 간의 차이가 선천적 기형 발생에 영향을 미쳤을 수 있습니다. 이 연구는 레지스트리의 방법론적 제한 사항(작은 표본 크기, 연구의 자발적 성격 및 무작위 배정되지 않은 설계 포함)으로 인해 아달리무맙과 주요 선천적 기형 사이에 연관성이 있는지 여부를 확실하게 밝힐 수 없습니다.
HUMIRA로 치료받은 IBD가 있는 임산부 10명을 대상으로 수행된 독립적인 임상 연구에서, 출생 당일에 모체 혈청뿐만 아니라 탯줄 혈액(n=10)과 영아 혈청(n=8)에서 아달리무맙 농도를 측정했습니다. HUMIRA의 마지막 투여는 분만 전 1일에서 56일 사이에 이루어졌습니다. 탯줄 혈액에서 아달리무맙 농도는 0.16-19.7 µg/mL, 영아 혈청에서 4.28-17.7 µg/mL, 모체 혈청에서 0-16.1 µg/mL였습니다. 한 가지 경우를 제외하고, 탯줄 혈액에서 아달리무맙 농도는 모체 혈청 농도보다 높았으며, 이는 아달리무맙이 태반을 통해 활발하게 이동함을 시사합니다. 또한, 한 영아는 다음과 같은 각 혈청 농도를 보였습니다. 6주(1.94 µg/mL), 7주(1.31 µg/mL), 8주(0.93 µg/mL) 및 11주(0.53 µg/mL). 이는 아달리무맙이 출생 후 최소 3개월 동안 자궁 내 노출된 영아의 혈청에서 검출될 수 있음을 시사합니다.
동물 데이터
배아-태아 출산 전후 발달 연구에서, 임신한 시험관 원숭이에게 임신 20일부터 97일까지 메토트렉세이트 없이 MRHD(모체 IV 용량 최대 100mg/kg/주)로 달성한 것의 최대 373배에 해당하는 노출을 유발하는 용량으로 아달리무맙을 투여했습니다(AUC 기준). 아달리무맙은 태아에게 해를 입히거나 기형을 유발하지 않았습니다.
8.2 수유
위험 요약
발표된 문헌의 사례 보고에서 얻은 제한적인 데이터는 모체 혈청 농도의 0.1%에서 1%에 해당하는 영아 용량으로 모유에서 아달리무맙이 검출된다는 것을 보여줍니다. 발표된 데이터에 따르면 아달리무맙은 큰 분자이며 위장관에서 분해되기 때문에 모유 수유를 하는 영아의 전신 노출은 낮을 것으로 예상됩니다. 그러나 위장관에서의 국소 노출의 영향은 알려져 있지 않습니다. 아달리무맙이 모유 수유를 하는 영아에게 부작용을 일으켰다는 보고는 없으며, 모유 생산에 영향을 미치지 않습니다. 모유 수유의 발달적 및 건강상 이점을 HUMIRA에 대한 모체의 임상적 필요와 HUMIRA 또는 기저 모체 질환으로 인해 모유 수유를 하는 영아에게 발생할 수 있는 잠재적 부작용과 함께 고려해야 합니다.
8.4 소아 사용
HUMIRA의 안전성과 유효성은 다음에 대해 확립되었습니다.
- 2세 이상 소아 환자에서 중등도에서 중증의 활동성 다관절 JIA의 징후 및 증상을 감소시키는 데 사용합니다.
- 6세 이상 소아 환자에서 중등도에서 중증의 활동성 크론병 치료에 사용합니다.
- 5세 이상 소아 환자에서 중등도에서 중증의 활동성 궤양성 대장염 치료에 사용합니다.
- 12세 이상 환자에서 중등도에서 중증의 털샘증 치료에 사용합니다.
- 2세 이상 소아 환자에서 비감염성 중간, 후방 및 전포도막염 치료에 사용합니다.
TNFα 억제 작용으로 인해 임신 중에 투여된 HUMIRA는 자궁 내 노출된 신생아 및 유아의 면역 반응에 영향을 미칠 수 있습니다. HUMIRA에 자궁 내 노출된 8명의 유아에 대한 데이터는 아달리무맙이 태반을 통과한다는 것을 시사합니다 [특정 인구 집단에서의 사용 (8.1)]. 유아에서 아달리무맙 농도가 높아진 것의 임상적 의미는 알려져 있지 않습니다. 노출된 유아에게 생백신 또는 생백신 약독화 백신을 투여하는 안전성은 알려져 있지 않습니다. 노출된 유아에게 백신 접종(생백신 또는 생백신 약독화 백신)을 하기 전에 위험과 이점을 고려해야 합니다.
HUMIRA를 포함한 TNF 억제제를 투여받은 어린이, 청소년 및 젊은 성인에서 림프종(간비장 T 세포 림프종 및 기타 악성 종양 포함)이 발생했다는 시판 후 보고가 있으며, 이 중 일부는 치명적이었습니다. [경고 및 주의 사항 (5.2)].
소아 특발성 관절염
JIA-I 연구에서 HUMIRA는 4세에서 17세 사이의 환자에서 활동성 다관절 JIA의 징후 및 증상을 감소시키는 것으로 나타났습니다. [임상 연구 (14.2)]. JIA-II 연구에서 2세에서 <4세 사이의 환자의 안전성 프로파일은 다관절 JIA가 있는 4세에서 17세 사이의 환자의 안전성 프로파일과 유사했습니다. [유해 반응 (6.1)]. HUMIRA는 2세 미만의 다관절 JIA 환자 또는 체중이 10kg 미만인 환자에게 연구되지 않았습니다.
다관절 JIA 시험에서 환자의 HUMIRA 안전성은 일반적으로 성인에서 관찰된 것과 유사했지만 몇 가지 예외가 있었습니다. [유해 반응 (6.1)].
2세 미만의 소아 환자에서 JIA에 대한 HUMIRA의 안전성 및 유효성은 확립되지 않았습니다.
소아 크론병
중등도에서 중증의 활동성 크론병 치료에 대한 HUMIRA의 안전성 및 유효성은 6세 이상 소아 환자에서 확립되었습니다. 이 적응증에 대한 HUMIRA의 사용은 성인에 대한 적절하고 잘 통제된 연구에서 얻은 증거와 192명의 소아 환자(6세에서 17세)에 대한 HUMIRA의 두 가지 용량 농도를 사용한 무작위 배정, 이중 맹검, 52주 임상 연구에서 얻은 추가 데이터에 의해 뒷받침됩니다. [유해 반응 (6.1), 임상 약리학 (12.2, 12.3), 임상 연구 (14.6)]. 6세에서 17세 사이의 환자의 유해 반응 프로파일은 성인과 유사했습니다.
6세 미만의 소아 환자에서 크론병에 대한 HUMIRA의 안전성 및 유효성은 확립되지 않았습니다.
소아 궤양성 대장염
중등도에서 중증의 활동성 궤양성 대장염 치료에 대한 HUMIRA의 안전성 및 유효성은 5세 이상 소아 환자에서 확립되었습니다. 이 적응증에 대한 HUMIRA의 사용은 성인에 대한 적절하고 잘 통제된 연구에서 얻은 증거와 93명의 소아 환자(5세에서 17세)에 대한 HUMIRA의 두 가지 용량 농도를 사용한 무작위 배정, 이중 맹검, 52주 임상 연구에서 얻은 추가 데이터에 의해 뒷받침됩니다. [유해 반응 (6.1), 임상 약리학 (12.3), 임상 연구 (14.8)]. 5세에서 17세 사이의 환자의 유해 반응 프로파일은 성인과 유사했습니다.
TNF 억제제에 대한 반응이 없거나 내약성이 없는 환자에서 HUMIRA의 유효성은 확립되지 않았습니다.
5세 미만의 소아 환자에서 궤양성 대장염에 대한 HUMIRA의 안전성 및 유효성은 확립되지 않았습니다.
소아 포도막염
비감염성 포도막염 치료에 대한 HUMIRA의 안전성 및 유효성은 2세 이상 소아 환자에서 확립되었습니다. HUMIRA의 사용은 성인에 대한 적절하고 잘 통제된 HUMIRA 연구와 90명의 소아 환자에 대한 2:1 무작위 배정, 대조군 임상 연구에서 얻은 증거에 의해 뒷받침됩니다. [임상 연구 (14.12)]. 2세 미만의 소아 환자에서 포도막염에 대한 HUMIRA의 안전성 및 유효성은 확립되지 않았습니다.
털샘증
HUMIRA는 12세 이상의 소아 환자에서 HS에 대한 사용이 성인 HS 환자에서 HUMIRA의 적절하고 잘 통제된 연구에서 얻은 증거에 의해 뒷받침됩니다. 추가적인 모집단 약동학적 모델링 및 시뮬레이션은 12세 이상의 소아 환자에서 체중 기반 HUMIRA 투여가 성인 HS 환자와 일반적으로 유사한 노출을 제공할 수 있음을 예측했습니다. HS의 경과는 성인 및 청소년 환자에서 충분히 유사하여 성인에서 청소년 환자로 데이터를 외삽할 수 있습니다. 12세 이상의 소아 환자에서 권장되는 용량은 체중을 기반으로 합니다. [용법 및 투여량 (2.6), 임상 약리 (12.3), 및 임상 연구 (14.10)].
HUMIRA의 안전성 및 유효성은 HS가 있는 12세 미만의 환자에서 확립되지 않았습니다.
8.5 노인 환자
RA-I에서 IV까지의 임상 연구에서 107명의 환자가 75세 이상인 것을 포함하여 65세 이상의 RA 환자 519명이 HUMIRA를 투여받았습니다. 이러한 환자와 젊은 환자 사이에 유효성에 대한 전반적인 차이가 관찰되지 않았습니다. HUMIRA를 투여받은 65세 이상의 환자에서 심각한 감염 및 악성 종양의 빈도가 65세 미만의 환자보다 높았습니다. 65세 이상의 환자에서 HUMIRA의 유익성과 위험을 고려하십시오. HUMIRA로 치료받는 환자에서 감염 또는 악성 종양의 발생을 면밀히 모니터링하십시오. [경고 및 주의 사항 (5.1, 5.2)].
10 과량투여
용량 제한 독성의 증거 없이 최대 10mg/kg의 용량이 임상 시험에서 환자에게 투여되었습니다. 과량 투여 시, 부작용이나 영향의 징후 또는 증상이 있는지 환자를 모니터링하고 적절한 증상 치료를 즉시 시작하는 것이 좋습니다.
11 설명
아달리무맙은 종양 괴사 인자 차단제입니다. 아달리무맙은 파지 디스플레이 기술을 사용하여 생성된 재조합 인간 IgG1 단일 클론 항체로, 인간에서 유래한 중쇄 및 경쇄 가변 영역과 인간 IgG1:k 불변 영역을 갖습니다. 아달리무맙은 포유 동물 세포(중국 햄스터 난소(CHO)) 발현 시스템에서 재조합 DNA 기술로 생산되며, 특정 바이러스 불활성화 및 제거 단계를 포함하는 공정으로 정제됩니다. 1330개의 아미노산으로 구성되어 있으며 분자량은 약 148킬로달톤입니다.
HUMIRA(아달리무맙) 주사제는 피하 주사용 무균 보존제 무첨가 용액으로 제공됩니다. 이 의약품은 단일 용량 사전 충전 펜(HUMIRA 펜), 단일 용량 1mL 사전 충전 유리 주사기 또는 단일 용량 기관용 바이알로 제공됩니다. 펜 안에는 단일 용량 1mL 사전 충전 유리 주사기가 들어 있습니다. HUMIRA 용액은 투명하고 무색이며 pH는 약 5.2입니다.
80mg/0.8mL 사전 충전 주사기 또는 사전 충전 펜은 0.8mL(80mg)의 의약품을 제공합니다. HUMIRA 0.8mL에는 아달리무맙(80mg), 만니톨(33.6mg), 폴리소르베이트 80(0.8mg) 및 주사용수, USP가 들어 있습니다.
40mg/0.4mL 사전 충전 주사기 또는 사전 충전 펜은 0.4mL(40mg)의 의약품을 제공합니다. HUMIRA 0.4mL에는 아달리무맙(40mg), 만니톨(16.8mg), 폴리소르베이트 80(0.4mg) 및 주사용수, USP가 들어 있습니다.
40mg/0.8mL 사전 충전 주사기, 사전 충전 펜 또는 단일 용량 기관용 바이알은 0.8mL(40mg)의 의약품을 제공합니다. HUMIRA 0.8mL에는 아달리무맙(40mg), 구연산 1수화물(1.04mg), 이수소 나트륨 인산 2수화물(1.22mg), 만니톨(9.6mg), 일수소 나트륨 인산 2수화물(0.69mg), 폴리소르베이트 80(0.8mg), 염화나트륨(4.93mg), 구연산나트륨(0.24mg) 및 주사용수, USP가 들어 있습니다. pH 조절을 위해 필요에 따라 수산화나트륨이 첨가됩니다.
20mg/0.2mL 사전 충전 주사기는 0.2mL(20mg)의 의약품을 제공합니다. HUMIRA 0.2mL에는 아달리무맙(20mg), 만니톨(8.4mg), 폴리소르베이트 80(0.2mg) 및 주사용수, USP가 들어 있습니다.
20mg/0.4mL 사전 충전 주사기는 0.4mL(20mg)의 의약품을 제공합니다. HUMIRA 0.4mL에는 아달리무맙(20mg), 구연산 1수화물(0.52mg), 이수소 나트륨 인산 2수화물(0.61mg), 만니톨(4.8mg), 일수소 나트륨 인산 2수화물(0.34mg), 폴리소르베이트 80(0.4mg), 염화나트륨(2.47mg), 구연산나트륨(0.12mg) 및 주사용수, USP가 들어 있습니다. pH 조절을 위해 필요에 따라 수산화나트륨이 첨가됩니다.
10mg/0.1mL 사전 충전 주사기는 0.1mL(10mg)의 의약품을 제공합니다. HUMIRA 0.1mL에는 아달리무맙(10mg), 만니톨(4.2mg), 폴리소르베이트 80(0.1mg) 및 주사용수, USP가 들어 있습니다.
10mg/0.2mL 사전 충전 주사기는 0.2mL(10mg)의 의약품을 제공합니다. HUMIRA 0.2mL에는 아달리무맙(10mg), 구연산 1수화물(0.26mg), 이수소 나트륨 인산 2수화물(0.31mg), 만니톨(2.4mg), 일수소 나트륨 인산 2수화물(0.17mg), 폴리소르베이트 80(0.2mg), 염화나트륨(1.23mg), 구연산나트륨(0.06mg) 및 주사용수, USP가 들어 있습니다. pH 조절을 위해 필요에 따라 수산화나트륨이 첨가됩니다.
12. 임상 약리
12.1 작용 기전
Adalimumab은 TNF-alpha에 특이적으로 결합하여 p55 및 p75 세포 표면 TNF 수용체와의 상호 작용을 차단합니다. 또한 Adalimumab은 보체 존재 하에 체외에서 표면 TNF 발현 세포를 용해합니다. Adalimumab은 림프독소(TNF-beta)를 결합하거나 비활성화하지 않습니다. TNF는 정상적인 염증 및 면역 반응에 관여하는 자연 발생 사이토카인입니다. TNF 농도 상승은 RA, JIA, PsA 및 AS 환자의 활액에서 발견되며 이러한 질환의 특징인 병적 염증 및 관절 파괴 모두에서 중요한 역할을 합니다. TNF 농도 증가는 건선 플라크에서도 발견됩니다. Ps에서 HUMIRA로 치료하면 표피 두께와 염증 세포의 침윤이 감소할 수 있습니다. 이러한 약력학적 활성과 HUMIRA가 임상 효과를 발휘하는 기전 간의 관계는 알려져 있지 않습니다.
또한 Adalimumab은 백혈구 이동을 담당하는 부착 분자(IC50 1-2 X 10-10M의 ELAM-1, VCAM-1 및 ICAM-1)의 농도 변화를 포함하여 TNF에 의해 유도되거나 조절되는 생물학적 반응을 조절합니다.
12.2 약력학
HUMIRA로 치료한 후 류마티스 관절염 환자에서 기준치에 비해 염증의 급성기 반응물(C-반응성 단백질 [CRP] 및 적혈구 침강 속도 [ESR]) 및 혈청 사이토카인(IL-6)의 농도가 감소했습니다. 크론병, 궤양성 대장염 및 화농성 한선염 환자에서도 CRP 농도 감소가 관찰되었습니다. 연골 파괴를 담당하는 조직 리모델링을 생성하는 기질 금속단백질분해효소(MMP-1 및 MMP-3)의 혈청 농도도 HUMIRA 투여 후 감소했습니다.
궤양성 대장염이 있는 5~17세 소아 환자의 경우 권장 HUMIRA 용량은 모델링된 용량/노출-효능 관계 및 약동학 데이터를 기반으로 합니다. 임상 시험(PUC-I 연구의 0~52주)에서 투여된 연구된 고용량과 권장 용량 간에 임상적으로 유의미한 효능 차이는 예상되지 않습니다. [14.8] [2.4].
12.3 약동학
Adalimumab의 약동학은 단일 정맥 투여 후 0.5~10mg/kg의 용량 범위에서 선형적이었습니다(HUMIRA는 정맥 투여용으로 승인되지 않았습니다). 20, 40, 80mg을 격주 및 매주 피하 투여한 후 RA 환자에서 adalimumab의 평균 혈청 최저 농도는 용량에 따라 거의 비례하여 증가했습니다. 평균 말기 반감기는 약 2주였으며 연구에 따라 10~20일이었습니다. 건강한 피험자와 RA 환자는 유사한 adalimumab 약동학을 보였습니다.
80mg을 격주로 투여받은 환자의 Adalimumab 노출은 40mg을 매주 투여받은 환자의 노출과 비슷한 것으로 추정됩니다.
흡수
단일 40mg 피하 투여 후 adalimumab의 평균 절대 생체 이용률은 64%였습니다. 건강한 피험자에게 40mg의 HUMIRA를 단일 피하 투여한 후 최대 농도에 도달하는 평균 시간은 5.5일(131±56시간)이었고 최대 혈청 농도는 4.7±1.6mcg/mL였습니다.
분포
RA 환자에서 0.25~10mg/kg 범위의 용량을 정맥 투여한 후 분포 용적(Vss)은 4.7~6.0L였습니다.
제거
RA 환자에서 adalimumab의 단일 용량 약동학은 0.25~10mg/kg 범위의 정맥 용량을 사용한 여러 연구에서 결정되었습니다. Adalimumab의 전신 청소율은 약 12mL/시간입니다. 2년 이상 투여하는 장기 연구에서 RA 환자에서 시간이 지남에 따라 청소율이 변한다는 증거는 없었습니다.
환자 집단
류마티스 관절염 및 강직성 척추염: HUMIRA 40mg을 격주로 투여받은 환자에서 adalimumab의 평균 항정 상태 최저 농도는 MTX 병용 치료 없이 약 5mcg/mL, MTX 병용 치료 시 약 8~9mcg/mL였습니다. 류마티스 관절염 환자 5명의 활액에서 adalimumab 농도는 혈청 농도의 31~96%였습니다. AS 환자에서 adalimumab의 약동학은 RA 환자의 약동학과 유사했습니다.
건선성 관절염: 40mg을 격주로 투여받은 환자에서 adalimumab의 평균 항정 상태 최저 농도는 MTX 병용 치료 없이 6~10mcg/mL, MTX 병용 치료 시 8.5~12mcg/mL였습니다.
판상 건선: HUMIRA 40mg을 격주로 치료하는 동안 adalimumab의 평균 항정 상태 최저 농도는 약 5~6mcg/mL였습니다.
성인 포도막염: HUMIRA 40mg을 격주로 치료하는 동안 adalimumab의 평균 항정 농도는 약 8~10mcg/mL였습니다.
성인 화농성 한선염: 0주차에 160mg을 투여받고 2주차에 80mg을 투여받은 후 2주차와 4주차에 adalimumab 최저 농도는 각각 약 7~8mcg/mL였습니다. HUMIRA 40mg을 매주 투여하는 동안 12주차부터 36주차까지의 평균 항정 상태 최저 농도는 약 7~11mcg/mL였습니다.
성인 크론병: 0주차에 160mg을 투여받고 2주차에 80mg을 투여받은 후 2주차와 4주차에 adalimumab의 평균 최저 농도는 약 12mcg/mL였습니다. HUMIRA 40mg을 격주로 투여하는 동안 24주차와 56주차의 평균 항정 상태 최저 농도는 7mcg/mL였습니다.
성인 궤양성 대장염: 0주차에 160mg을 투여받고 2주차에 80mg을 투여받은 후 2주차와 4주차에 adalimumab의 평균 최저 농도는 약 12mcg/mL였습니다. HUMIRA 40mg을 격주 및 매주 투여한 후 52주차의 평균 항정 상태 최저 농도는 각각 약 8mcg/mL 및 15mcg/mL였습니다.
약동학에 대한 항의약품 항체 효과
류마티스 관절염: 항-아달리무맙 항체가 있는 경우 아달리무맙의 외관상 청소율이 더 높아지는 경향이 확인되었습니다.
소아 궤양성 대장염: ECL 분석법으로 측정한 아달리무맙에 대한 항체는 중등도에서 중증의 활동성 궤양성 대장염 소아 환자에서 혈청 아달리무맙 농도 감소와 관련이 있었습니다.
화농성 한선염: 중등도에서 중증의 HS 환자에서 아달리무맙에 대한 항체는 혈청 아달리무맙 농도 감소와 관련이 있었습니다. 일반적으로 아달리무맙에 대한 항체 역가가 증가함에 따라 혈청 아달리무맙 농도 감소 정도가 더 커집니다.
특정 집단
노인 환자: 40세에서 >75세 사이의 RA 환자에서 연령이 증가함에 따라 청소율이 감소하는 것이 관찰되었습니다.
소아 환자:
다관절형 소아 특발성 관절염:
- 4 세 ~ 17세: 아달리무맙 평균 정상 상태 최저 농도는 2주마다 20mg HUMIRA를 피하 주사로 단독 요법 또는 MTX 병용 요법으로 투여받은 체중 <30kg 환자에서 각각 6.8mcg/mL 및 10.9mcg/mL였습니다. 아달리무맙 평균 정상 상태 최저 농도는 2주마다 40mg HUMIRA를 피하 주사로 단독 요법 또는 MTX 병용 요법으로 투여받은 체중 ≥30kg 환자에서 각각 6.6mcg/mL 및 8.1mcg/mL였습니다.
- 2 세 ~ <4세 또는 4세 이상이고 체중이 <15kg인 환자: 아달리무맙 평균 정상 상태 최저 아달리무맙 농도는 2주마다 HUMIRA를 피하 주사로 단독 요법 또는 MTX 병용 요법으로 투여받은 환자에서 각각 6.0mcg/mL 및 7.9mcg/mL였습니다.
소아 화농성 한선염: 권장 복용량으로 투여받은 HS 청소년 환자의 아달리무맙 농도는 집단 약동학 모델링 및 시뮬레이션을 기반으로 HS 성인 환자에서 관찰된 것과 유사할 것으로 예상됩니다.
소아 크론병: 아달리무맙 평균 ± SD 농도는 체중이 ≥ 40kg인 환자에서 0주차에 160mg, 2주차에 80mg을 투여한 후 4주차에 15.7±6.5mcg/mL였고, 2주마다 40mg을 투여한 후 52주차에 10.5±6.0mcg/mL였습니다. 아달리무맙 평균 ± SD 농도는 체중이 < 40kg인 환자에서 0주차에 80mg, 2주차에 40mg을 투여한 후 4주차에 10.6±6.1mcg/mL였고, 2주마다 20mg을 투여한 후 52주차에 6.9±3.6mcg/mL였습니다.
소아 궤양성 대장염: 아달리무맙 평균 정상 상태 최저 농도는 5세에서 17세 사이의 소아 UC 환자에서 2주마다 0.6mg/kg(최대 40mg)을 피하 투여한 후 52주차에 5.0±3.3mcg/mL였습니다. 매주 0.6mg/kg(최대 40mg)을 투여받은 환자의 경우 5세에서 17세 사이의 소아 UC 환자에서 평균 정상 상태 최저 농도는 52주차에 15.7±5.6mcg/mL였습니다.
남성 및 여성 환자: 환자의 체중을 보정한 후 성별 관련 약동학적 차이는 관찰되지 않았습니다. 건강한 피험자와 류마티스 관절염 환자는 유사한 아달리무맙 약동학을 보였습니다.
신장 또는 간 장애 환자: 간 또는 신장 장애 환자에 대한 약동학적 데이터는 없습니다.
류마티스 인자 또는 CRP 농도: 권장 용량보다 낮은 용량을 투여받은 RA 환자와 류마티스 인자 또는 CRP 농도가 높은 RA 환자에서 외관상 청소율이 약간 증가한 것으로 예측되었습니다. 이러한 증가는 임상적으로 중요하지 않을 가능성이 높습니다.
약물 상호 작용 연구:
Methotrexate: MTX는 RA 환자에서 단회 및 다회 투여 후 아달리무맙 외관상 청소율을 각각 29% 및 44% 감소시켰습니다. [약물 상호 작용 (7.1) 참조].
13 비임상 독성학
14. 임상 연구
14.1 류마티스 관절염
HUMIRA의 효능 및 안전성은 미국 류마티스 학회(ACR) 기준에 따라 진단된 활동성 류마티스 관절염(RA)이 있는 18세 이상의 환자를 대상으로 한 5건의 무작위배정, 이중맹검 연구에서 평가되었습니다. 환자들은 최소 6개의 부은 관절과 9개의 압통 관절을 가지고 있었습니다. HUMIRA는 메토트렉세이트(MTX)(12.5~25mg, RA-I, RA-III 및 RA-V 연구)와 병용하거나 단독 요법(RA-II 및 RA-V 연구)으로 또는 다른 질병 조절 항류마티스제(DMARD)(RA-IV 연구)와 병용하여 피하 투여되었습니다.
RA-I 연구에서는 최소 1개 이상, 최대 4개의 DMARD로 치료에 실패했고 MTX에 대한 반응이 불충분한 271명의 환자를 평가했습니다. 20, 40 또는 80mg의 HUMIRA 또는 위약을 24주 동안 격주로 투여했습니다.
RA-II 연구에서는 최소 1개의 DMARD로 치료에 실패한 544명의 환자를 평가했습니다. 위약, 20 또는 40mg의 HUMIRA를 26주 동안 격주 또는 매주 단독 요법으로 투여했습니다.
RA-III 연구에서는 MTX에 대한 반응이 불충분한 619명의 환자를 평가했습니다. 환자들은 최대 52주 동안 위약, 격주로 40mg의 HUMIRA와 대체 주에 위약 주사 또는 매주 20mg의 HUMIRA를 투여받았습니다. RA-III 연구는 52주차에 질병 진행 억제(X선 결과로 감지됨)라는 추가적인 1차 평가 변수를 가졌습니다. 처음 52주가 완료되면 457명의 환자가 공개 라벨 연장 연구에 등록하여 최대 5년 동안 격주로 40mg의 HUMIRA를 투여받았습니다.
RA-IV 연구에서는 DMARD-naïve이거나 최소 28일 동안 치료가 안정적인 경우 기존의 류마티스 치료를 계속할 수 있는 636명의 환자를 대상으로 안전성을 평가했습니다. 환자들은 24주 동안 격주로 40mg의 HUMIRA 또는 위약을 무작위로 투여받았습니다.
RA-V 연구에서는 18세 이상이고 MTX-naïve이며 기간이 3년 미만인 중등도에서 중증의 활동성 RA 환자 799명을 평가했습니다. 환자들은 104주 동안 MTX(8주차까지 20mg/주까지 최적화됨), 격주로 HUMIRA 40mg 또는 HUMIRA/MTX 병용 요법을 무작위로 투여받았습니다. 환자들은 징후 및 증상과 관절 손상의 방사선학적 진행에 대해 평가되었습니다. 연구에 등록된 환자의 평균 질병 기간은 5개월이었습니다. 달성된 평균 MTX 용량은 20mg이었습니다.
임상 반응
RA-II 및 III 연구에서 ACR 20, 50 및 70 반응을 달성한 HUMIRA 치료 환자의 비율은 표 3에 나와 있습니다.
| RA-II 연구 단독 요법 (26주) |
RA-III 연구 메토트렉세이트 병용 (24주 및 52주) |
||||
| 반응 | 위약 | HUMIRA | HUMIRA | 위약/MTX | HUMIRA/MTX |
| 40mg 격주 | 40mg 매주 | 40mg 격주 | |||
| 투여 | 투여 | ||||
| N=110 | N=113 | N=103 | N=200 | N=207 | |
| ACR20 | |||||
| 6개월 | 19% | 46%* | 53%* | 30% | 63%* |
| 12개월 | NA | NA | NA | 24% | 59%* |
| ACR50 | |||||
| 6개월 | 8% | 22%* | 35%* | 10% | 39%* |
| 12개월 | NA | NA | NA | 10% | 42%* |
| ACR70 | |||||
| 6개월 | 2% | 12%* | 18%* | 3% | 21%* |
| 12개월 | NA | NA | NA | 5% | 23%* |
| * p<0.01, HUMIRA vs. placebo | |||||
RA-I 연구 결과는 RA-III 연구 결과와 유사했습니다. RA-I 연구에서 격주로 HUMIRA 40mg을 투여받은 환자는 6개월 시점에서 ACR 20, 50, 70 반응률이 각각 65%, 52%, 24%였으며, 이는 위약군의 반응률인 13%, 7%, 3%와 비교했을 때 유의미한 차이를 보였습니다(p<0.01).
RA-II 및 RA-III 연구에 대한 ACR 반응 기준 구성 요소의 결과는 표 4에 나와 있습니다. ACR 반응률 및 ACR 반응의 모든 구성 요소에서의 개선은 104주까지 유지되었습니다. RA-III 연구에서 2년 동안 격주로 HUMIRA 40mg을 투여받은 환자의 20%가 주요 임상 반응(6개월 동안 ACR 70 반응 유지로 정의됨)을 달성했습니다. RA-III 연구의 공개 라벨 부분에서 HUMIRA를 지속적으로 투여받은 환자의 경우 유사한 비율의 환자에서 ACR 반응이 최대 5년 동안 유지되었습니다.
| RA-II 연구 | RA-III 연구 | |||||||
| 매개변수(중앙값) | 위약 N=110 |
HUMIRAa N=113 |
위약/MTX N=200 |
HUMIRAa/MTX N=207 |
||||
| 기준치 | 26주차 | 기준치 | 26주차 | 기준치 | 24주차 | 기준치 | 24주차 | |
| 압통 관절 수(0-68) | 35 | 26 | 31 | 16* | 26 | 15 | 24 | 8* |
| 종창 관절 수(0-66) | 19 | 16 | 18 | 10* | 17 | 11 | 18 | 5* |
| 의사의 전체 평가b | 7.0 | 6.1 | 6.6 | 3.7* | 6.3 | 3.5 | 6.5 | 2.0* |
| 환자의 전체 평가b | 7.5 | 6.3 | 7.5 | 4.5* | 5.4 | 3.9 | 5.2 | 2.0* |
| 통증b | 7.3 | 6.1 | 7.3 | 4.1* | 6.0 | 3.8 | 5.8 | 2.1* |
| 장애 지수(HAQ)c | 2.0 | 1.9 | 1.9 | 1.5* | 1.5 | 1.3 | 1.5 | 0.8* |
| CRP(mg/dL) | 3.9 | 4.3 | 4.6 | 1.8* | 1.0 | 0.9 | 1.0 | 0.4* |
| a 격주로 투여된 HUMIRA 40mg b 시각 아날로그 척도, 0 = 최상, 10 = 최악 c 건강 평가 설문지의 장애 지수, 0 = 최상, 3 = 최악, 옷 입기/몸단장하기, 일어나기, 식사하기, 걷기, 뻗기, 잡기, 위생 유지, 일상 활동 유지와 같은 활동을 수행하는 환자의 능력을 측정함 * p<0.001, HUMIRA vs. 위약, 기준치 대비 평균 변화를 기준으로 함 |
||||||||
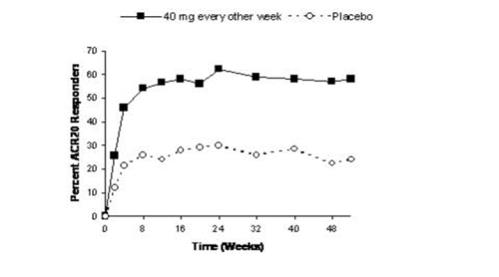
ACR 20 반응에 대한 연구 RA-III의 시간 경과는 그림 1에 나와 있습니다.
연구 RA-III에서 24주차에 ACR 20 반응을 보인 환자의 85%가 52주차까지 반응을 유지했습니다. 연구 RA-I 및 연구 RA-II의 ACR 20 반응에 대한 시간 경과도 유사했습니다.
그림 1. 52주 동안의 연구 RA-III ACR 20 반응
연구 RA-IV에서 HUMIRA 40mg을 격주로 표준 치료와 함께 투여받은 환자의 53%가 24주차에 ACR 20 반응을 보인 반면, 위약과 표준 치료를 병행한 환자는 35%였습니다(p<0.001). HUMIRA(adalimumab)와 다른 DMARD의 병용과 관련된 특이적인 이상 반응은 관찰되지 않았습니다.
최근에 RA가 발병한 MTX 치료 경험이 없는 환자를 대상으로 한 연구 RA-V에서 HUMIRA와 MTX 병용 요법은 52주차에 MTX 단독 요법이나 HUMIRA 단독 요법보다 ACR 반응을 달성한 환자의 비율이 더 높았으며, 104주차까지 반응이 유지되었습니다(표 5 참조).
| 반응 | MTXb N=257 |
HUMIRAc N=274 |
HUMIRA/MTX N=268 |
| ACR20 52주차 104주차 |
63% 56% |
54% 49% |
73% 69% |
| ACR50 52주차 104주차 |
46% 43% |
41% 37% |
62% 59% |
| ACR70 52주차 104주차 |
27% 28% |
26% 28% |
46% 47% |
| 주요 임상 반응 a | 28% | 25% | 49% |
| a 주요 임상 반응은 6개월 연속 ACR70 반응을 달성한 것으로 정의됩니다. b p<0.05, HUMIRA/MTX vs. MTX(ACR 20) p<0.001, HUMIRA/MTX vs. MTX(ACR 50 및 70, 주요 임상 반응) c p<0.001, HUMIRA/MTX vs. HUMIRA |
|||
52주차에 연구 RA-V에 대한 ACR 반응 기준의 모든 개별 구성 요소가 HUMIRA/MTX 그룹에서 개선되었으며, 개선 사항은 104주차까지 유지되었습니다.
방사선학적 반응
연구 RA-III에서 구조적 관절 손상을 방사선학적으로 평가했으며, 기준치와 비교하여 12개월째에 총 Sharp 점수(TSS)의 변화와 그 구성 요소인 미란 점수 및 관절 간격 협착(JSN) 점수로 나타냈습니다. 기준치에서 중앙값 TSS는 위약 그룹과 40mg 격주 그룹에서 각각 약 55였습니다. 결과는 표 6에 나와 있습니다. HUMIRA/MTX 치료를 받은 환자는 52주차에 MTX만 투여받은 환자보다 방사선학적 진행이 적었습니다.
| 위약/MTX | HUMIRA/MTX 40mg 격주 투여 |
위약/MTX- HUMIRA/MTX(95% 신뢰 구간*) |
P-값** | |
| 총 Sharp 점수 | 2.7 | 0.1 | 2.6(1.4, 3.8) | <0.001 |
| 미란 점수 | 1.6 | 0.0 | 1.6(0.9, 2.2) | <0.001 |
| JSN 점수 | 1.0 | 0.1 | 0.9(0.3, 1.4) | 0.002 |
| *MTX와 HUMIRA 간의 변화 점수 차이에 대한 95% 신뢰 구간. **순위 분석 기반 |
||||
개방형 연장 연구 RA-III에서 모든 HUMIRA 용량으로 치료받은 원래 환자의 77%가 2년째에 방사선학적 평가를 받았습니다. TSS로 측정한 바와 같이 환자들은 구조적 손상의 억제를 유지했습니다. 54%는 TSS의 변화가 0 이하로 정의된 구조적 손상의 진행이 없었습니다. 40mg HUMIRA로 격주로 치료받은 원래 환자의 55%가 5년째에 방사선학적 평가를 받았습니다. 환자들은 TSS의 변화가 0 이하로 정의된 구조적 손상의 진행이 없는 경우가 50%로 구조적 손상의 지속적인 억제를 보였습니다.
연구 RA-V에서 구조적 관절 손상은 연구 RA-III에서와 같이 평가되었습니다. TSS, 미란 점수 및 JSN의 변화로 평가한 바와 같이 HUMIRA/MTX 병용 요법 그룹에서 MTX 또는 HUMIRA 단독 요법 그룹에 비해 52주차뿐만 아니라 104주차에도 방사선학적 진행의 더 큰 억제가 관찰되었습니다(표 7 참조).
| MTXa N=257 |
HUMIRAa,b N=274 |
HUMIRA/MTX N=268 |
||
| 52주차 | 총 Sharp 점수 | 5.7 (4.2, 7.3) | 3.0 (1.7, 4.3) | 1.3 (0.5, 2.1) |
| 미란 점수 | 3.7 (2.7, 4.8) | 1.7 (1.0, 2.4) | 0.8 (0.4, 1.2) | |
| JSN 점수 | 2.0 (1.2, 2.8) | 1.3 (0.5, 2.1) | 0.5 (0.0, 1.0) | |
| 104주차 | 총 Sharp 점수 | 10.4 (7.7, 13.2) | 5.5 (3.6, 7.4) | 1.9 (0.9, 2.9) |
| 미란 점수 | 6.4 (4.6, 8.2) | 3.0 (2.0, 4.0) | 1.0 (0.4, 1.6) | |
| JSN 점수 | 4.1 (2.7, 5.4) | 2.6 (1.5, 3.7) | 0.9 (0.3, 1.5) | |
| * 평균(95% 신뢰 구간) a p<0.001, HUMIRA/MTX vs. 52주 및 104주차의 MTX 및 104주차의 HUMIRA/MTX vs. HUMIRA b p<0.01, 52주차의 HUMIRA/MTX vs. HUMIRA |
||||
신체 기능 반응
RA-I에서 IV까지의 연구에서, HUMIRA는 연구 시작 시점부터 연구 종료 시점까지의 Health Assessment Questionnaire (HAQ-DI) 장애 지수에서 위약보다 유의미하게 더 큰 개선을 보였으며, The Short Form Health Survey (SF 36)로 평가한 건강 결과에서 위약보다 유의미하게 더 큰 개선을 보였습니다. 개선은 신체 구성 요약(PCS)과 정신 구성 요약(MCS) 모두에서 나타났습니다.
RA-III 연구에서, 52주차에 HAQ-DI의 기준치 대비 평균(95% CI) 개선은 HUMIRA 환자의 경우 0.60(0.55, 0.65)였고, 위약/MTX(p<0.001) 환자의 경우 0.25(0.17, 0.33)였습니다. HUMIRA 치료를 받은 환자의 63%가 연구의 이중맹검 부분에서 52주차에 HAQ-DI에서 0.5 이상 개선되었습니다. 이러한 환자의 82%가 104주차까지 개선을 유지했으며, 유사한 비율의 환자가 260주(5년)의 공개 라벨 치료를 통해 이러한 반응을 유지했습니다. SF-36의 평균 개선은 156주차(3년)에 측정이 끝날 때까지 유지되었습니다.
RA-V 연구에서, HAQ-DI 및 SF-36의 신체 구성 요소는 52주차에 HUMIRA/MTX 병용 요법 그룹이 MTX 단독 요법 그룹 또는 HUMIRA 단독 요법 그룹보다 더 큰 개선(p<0.001)을 보였으며, 이는 104주차까지 유지되었습니다.
14.2 소아 특발성 관절염
활동성 다관절 소아 특발성 관절염(JIA) 환자를 대상으로 한 두 건의 연구(JIA-I 연구 및 JIA-II 연구)에서 HUMIRA의 안전성 및 유효성을 평가했습니다.
JIA-I 연구
다관절 JIA를 앓고 있는 4~17세 환자 171명을 대상으로 한 다기관, 무작위 배정, 중단, 이중맹검, 병행 그룹 연구에서 HUMIRA의 안전성 및 유효성을 평가했습니다. 연구에서 환자는 MTX 치료 그룹 또는 비 MTX 치료 그룹의 두 그룹으로 계층화되었습니다. 모든 환자는 NSAID, 진통제, 코르티코스테로이드 또는 DMARD로 이전에 치료를 받았음에도 불구하고 활동성 중등도 또는 중증 질환의 징후를 보여야 했습니다. 이전에 생물학적 DMARD로 치료를 받은 환자는 연구에서 제외되었습니다.
이 연구는 공개 라벨 선도 단계(OL-LI, 16주), 이중맹검 무작위 중단 단계(DB, 32주), 공개 라벨 연장 단계(OLE-BSA, 최대 136주) 및 공개 라벨 고정 용량 단계(OLE-FD, 16주)의 네 단계로 구성되었습니다. 연구의 처음 세 단계에서 HUMIRA는 체표면적을 기준으로 24mg/m2의 용량으로 최대 총 체중 용량 40mg을 격주로 피하(SC) 투여했습니다. OLE-FD 단계에서 환자의 체중이 30kg 미만인 경우 격주로 HUMIRA 20mg을 SC로 투여받았고, 체중이 30kg 이상인 경우 격주로 HUMIRA 40mg을 SC로 투여받았습니다. 환자들은 안정적인 용량의 NSAID 및/또는 프레드니손(≤0.2mg/kg/일 또는 최대 10mg/일)을 계속 투여받았습니다.
OL-LI 단계 종료 시점에 소아 ACR 30 반응을 보인 환자는 연구의 이중맹검(DB) 단계에 무작위 배정되었고 32주 동안 또는 질병 재발 시까지 격주로 HUMIRA 또는 위약을 투여받았습니다. 질병 재발은 6가지 소아 ACR 핵심 기준 중 ≥3가지에서 기준치보다 ≥30% 악화, ≥2개의 활성 관절 및 6가지 기준 중 1개 이하에서 30% 초과 개선으로 정의되었습니다. 32주 후 또는 DB 단계 동안 질병 재발 시점에 환자는 체중에 따라 고정 용량 요법(OLE-FD 단계)으로 전환하기 전에 BSA 요법(OLE-BSA)을 기반으로 공개 라벨 연장 단계에서 치료를 받았습니다.
JIA-I 연구 임상 반응
16주 OL-LI 단계 종료 시점에 MTX 계층 환자의 94%와 비 MTX 계층 환자의 74%가 소아 ACR 30 반응을 보였습니다. DB 단계에서 HUMIRA를 투여받은 환자는 위약을 투여받은 환자에 비해 MTX를 투여하지 않은 경우(43% vs. 71%)와 MTX를 투여한 경우(37% vs. 65%) 모두 질병 재발을 경험한 환자가 유의미하게 적었습니다. HUMIRA로 치료받은 환자는 위약으로 치료받은 환자에 비해 48주차에 소아 ACR 30/50/70 반응을 지속적으로 보인 환자가 더 많았습니다. 소아 ACR 반응은 연구 전반에 걸쳐 HUMIRA를 투여받은 환자에서 OLE 단계에서 최대 2년 동안 유지되었습니다.
JIA-II 연구
중등도에서 중증의 활동성 다관절 JIA를 앓고 있는 2~<4세 또는 4세 이상 15kg 미만의 환자 32명을 대상으로 한 공개 라벨 다기관 연구에서 HUMIRA를 평가했습니다. 대부분의 환자(97%)는 최대 120주 동안 격주로 24mg/m2(최대 20mg) 용량의 HUMIRA를 1회 SC 주사로 24주 이상 투여받았습니다. 연구 중에 대부분의 환자는 MTX를 병용 투여받았으며, 코르티코스테로이드 또는 NSAID를 사용했다고 보고한 환자는 더 적었습니다. 이 연구의 주요 목표는 안전성을 평가하는 것이었습니다[이상반응 (6.1) 참조].
14.3 건선성 관절염
건선성 관절염(PsA) 환자 413명을 대상으로 한 두 건의 무작위 배정, 이중맹검, 위약 대조 연구에서 HUMIRA의 안전성 및 유효성을 평가했습니다. 두 연구를 완료한 후 383명의 환자가 공개 라벨 연장 연구에 등록했으며, 이 연구에서 HUMIRA 40mg을 격주로 투여했습니다.
PsA-I 연구에는 다음 형태 중 하나에서 NSAID 요법에 대한 반응이 불충분한 중등도에서 중증의 활동성 PsA(>3개의 부은 관절 및 >3개의 압통 관절)를 가진 성인 환자 313명이 등록했습니다. (1) 원위 지골간(DIP) 침범(N=23), (2) 다관절염(류마티스 결절 없음 및 판상 건선 존재)(N=210), (3) 절단성 관절염(N=1), (4) 비대칭성 PsA(N=77) 또는 (5) AS 유사(N=2). 등록 시 MTX 요법(>1개월 동안 ≤30mg/주의 안정 용량)을 받고 있던 환자(313명 중 158명)는 동일한 용량의 MTX를 계속 투여받을 수 있었습니다. 연구의 24주 이중맹검 기간 동안 HUMIRA 40mg 또는 위약을 격주로 투여했습니다.
위약과 비교했을 때 HUMIRA 치료는 질병 활성도 측정에서 개선을 보였습니다(표 8 및 9 참조). HUMIRA를 투여받은 PsA 환자 중 일부 환자에서는 첫 번째 방문(2주) 시점에 임상 반응이 나타났으며, 진행 중인 공개 라벨 연구에서 최대 88주까지 유지되었습니다. 절단성 관절염 및 강직성 척추염 유사 아형을 가진 환자는 소수만 등록되었지만, 각 건선성 관절염 아형을 가진 환자에서 유사한 반응이 나타났습니다. 기준치에서 MTX 요법을 병용 투여받았는지 여부와 관계없이 반응은 유사했습니다.
신체 표면적(BSA)의 최소 3% 이상이 건선에 이환된 환자들을 대상으로 건선 부위 중증도 지수(PASI) 반응을 평가했습니다. 24주차에 PASI가 75% 또는 90% 개선된 환자의 비율은 HUMIRA 군(N=69)에서 각각 59% 및 42%였던 반면, 위약 군(N=69)에서는 각각 1% 및 0%였습니다(p<0.001). PASI 반응은 일부 환자에서 첫 번째 방문(2주) 시 나타났습니다. 반응은 기준 시점에 병용 MTX 요법을 받았는지 여부와 관계없이 유사했습니다.
| 위약 N=162 |
HUMIRA* N=151 |
|
| ACR20 12주차 24주차 |
14% 15% |
58% 57% |
| ACR50 12주차 24주차 |
4% 6% |
36% 39% |
| ACR70 12주차 24주차 |
1% 1% |
20% 23% |
| * HUMIRA와 위약 비교에 대한 모든 p<0.001 | ||
| 위약 N=162 |
HUMIRA* N=151 |
|||
| 매개변수: 중앙값 | 기준선 | 24주차 | 기준선 | 24주차 |
| 압통 관절 수a | 23.0 | 17.0 | 20.0 | 5.0 |
| 종창 관절 수b | 11.0 | 9.0 | 11.0 | 3.0 |
| 의사의 종합 평가c | 53.0 | 49.0 | 55.0 | 16.0 |
| 환자의 종합 평가c | 49.5 | 49.0 | 48.0 | 20.0 |
| 통증c | 49.0 | 49.0 | 54.0 | 20.0 |
| 장애 지수(HAQ) d | 1.0 | 0.9 | 1.0 | 0.4 |
| CRP(mg/dL)e | 0.8 | 0.7 | 0.8 | 0.2 |
| * 중앙값 변화를 기준으로 HUMIRA 대 위약 비교에 대한 p<0.001 a 척도 0-78 b 척도 0-76 c 시각 아날로그 척도, 0=최상, 100=최악 d 건강 평가 설문지의 장애 지수, 0=최상, 3=최악, 옷 입기/몸단장하기, 일어서기, 식사하기, 걷기, 뻗기, 잡기, 위생 유지, 일상 활동 유지와 같은 활동을 수행하는 환자의 능력을 측정합니다. e 정상 범위: 0-0.287mg/dL |
||||
DMARD 치료에 대해 최적 이하의 반응을 보인 중등도에서 중증의 건선성 관절염 환자 100명을 대상으로 실시한 12주 추가 연구에서도 유사한 결과가 나타났으며, 이는 등록 시 ≥3개의 압통 관절과 ≥3개의 부종 관절로 나타났습니다.
방사선학적 반응
PsA 연구에서 방사선학적 변화를 평가했습니다. 손, 손목 및 발의 방사선 사진은 이중맹검 기간 동안 환자가 HUMIRA 또는 위약을 투여받은 기준점과 24주차, 그리고 모든 환자가 공개 라벨 HUMIRA를 투여받은 48주차에 촬영했습니다. 원위 지절간 관절을 포함하는 수정된 총 Sharp 점수(mTSS)(즉, 류마티스 관절염에 사용된 TSS와 동일하지 않음)를 사용하여 치료군을 모르는 판독자가 방사선 사진을 평가했습니다.
HUMIRA 치료를 받은 환자는 위약 치료를 받은 환자에 비해 방사선학적 진행 억제 효과가 더 컸으며, 이러한 효과는 48주차까지 유지되었습니다(표 10 참조).
| 위약 N=141 |
HUMIRA N=133 |
||
| 24주차 | 24주차 | 48주차 | |
| 기준 평균 | 22.1 | 23.4 | 23.4 |
| 평균 변화 ± SD | 0.9 ± 3.1 | -0.1 ± 1.7 | -0.2 ± 4.9* |
| * HUMIRA 48주차와 위약 24주차 간의 차이에 대한 <0.001(주요 분석) | |||
신체 기능 반응
PsA-I 연구에서, 신체 기능 및 장애는 HAQ 장애 지수(HAQ-DI) 및 SF-36 건강 설문지를 사용하여 평가되었습니다. 격주 간격으로 HUMIRA 40mg을 투여받은 환자는 위약군(12주 및 24주째에 각각 평균 1% 및 3% 감소)에 비해 HAQ-DI 점수에서 기준치 대비 더 큰 개선을 보였습니다(12주 및 24주째에 각각 평균 47% 및 49% 감소). 12주 및 24주째에 HUMIRA로 치료받은 환자는 위약으로 치료받은 환자에 비해 SF-36 신체 구성 요약 점수에서 기준치 대비 더 큰 개선을 보였으며, SF-36 정신 구성 요약 점수에서는 악화가 없었습니다. HAQ-DI를 기반으로 한 신체 기능의 개선은 연구의 공개 라벨 부분을 통해 최대 84주 동안 유지되었습니다.
14.4 강직성 척추염
HUMIRA 40mg의 격주 투여 안전성 및 유효성은 글루코코르티코이드, NSAID, 진통제, 메토트렉세이트 또는 설파살라진에 대한 반응이 불충분한 활동성 강직성 척추염(AS) 환자 315명을 대상으로 한 무작위배정, 24주, 이중맹검, 위약 대조 연구에서 평가되었습니다. 활동성 AS는 다음 세 가지 기준 중 두 가지 이상을 충족하는 환자로 정의되었습니다. (1) Bath AS 질병 활성도 지수(BASDAI) 점수 ≥4cm, (2) 총 요통에 대한 시각 아날로그 점수(VAS) ≥ 40mm, 및 (3) 아침 강직 ≥ 1시간. 맹검 기간 후에는 최대 28주 동안 격주 간격으로 HUMIRA 40mg을 피하 투여받는 공개 라벨 기간이 이어졌습니다.
질병 활성도 측정치의 개선은 2주차에 처음 관찰되었으며 그림 2와 표 11에서와 같이 24주까지 유지되었습니다.
전체 척추 강직이 있는 환자(n=11)의 반응은 전체 강직이 없는 환자와 유사했습니다.
그림 2. 방문별 ASAS 20 반응, AS-I 연구
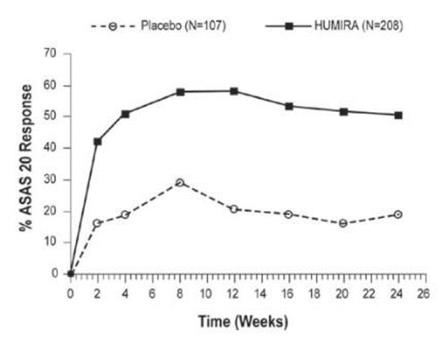
12주차에 ASAS 20/50/70 반응은 HUMIRA를 투여받은 환자의 각각 58%, 38%, 23%에서 달성되었으며, 위약을 투여받은 환자의 경우 각각 21%, 10%, 5%였습니다(p <0.001). 유사한 반응이 24주차에 나타났으며 최대 52주 동안 공개 라벨 HUMIRA를 투여받은 환자에서 유지되었습니다.
HUMIRA로 치료받은 환자의 비율이 더 높았으며(22%), 24주차에 낮은 수준의 질병 활성도(네 가지 ASAS 반응 매개변수 각각에서 [0~100mm 척도에서] <20의 값으로 정의됨)에 도달했습니다(위약군: 6%).
| 위약 N=107 |
HUMIRA N=208 |
|||
| 기준치 평균 |
24주차 평균 |
기준치 평균 |
24주차 평균 |
|
| ASAS 20 반응 기준* | ||||
| 환자의 질병 활성도에 대한 전반적 평가a* | 65 | 60 | 63 | 38 |
| 총 요통* | 67 | 58 | 65 | 37 |
| 염증b* | 6.7 | 5.6 | 6.7 | 3.6 |
| BASFIc* | 56 | 51 | 52 | 34 |
| BASDAId 점수* | 6.3 | 5.5 | 6.3 | 3.7 |
| BASMIe 점수* | 4.2 | 4.1 | 3.8 | 3.3 |
| Tragus to wall (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| 요추 굴곡(cm) | 4.1 | 4.0 | 4.2 | 4.4 |
| 경추 회전(도) | 42.2 | 42.1 | 48.4 | 51.6 |
| 요추 측면 굴곡(cm) | 8.9 | 9.0 | 9.7 | 11.7 |
| Intermalleolar distance (cm) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRPf* | 2.2 | 2.0 | 1.8 | 0.6 |
| a 0 = “없음” 및 100 = “심각함”인 시각 아날로그 척도(VAS)에서 측정했을 때 최소 20% 및 10단위 개선된 피험자 비율 b BASDAI 질문 5 및 6의 평균(‘d’에 정의됨) c Bath 강직성 척추염 기능 지수 d Bath 강직성 척추염 질병 활성도 지수 e Bath 강직성 척추염 계측 지수 f C-반응성 단백질(mg/dL) * 24주차에 HUMIRA와 위약을 비교했을 때 통계적으로 유의미함 |
||||
강직성 척추염 환자 82명을 대상으로 한 두 번째 무작위배정, 다기관, 이중맹검, 위약 대조 연구에서도 유사한 결과가 나타났습니다.
HUMIRA로 치료받은 환자는 24주차에 위약으로 치료받은 환자와 비교하여 강직성 척추염 삶의 질 설문지(ASQoL) 점수(-3.6 vs. -1.1) 및 단축형 건강 설문지(SF-36) 신체 구성 요약(PCS) 점수(7.4 vs. 1.9)에서 기준치 대비 개선을 달성했습니다.
14.5 성인 크론병
무작위배정, 이중맹검, 위약 대조 연구에서 중등도~중증 활동성 크론병(CD)(크론병 활성도 지수(CDAI) ≥ 220 및 ≤ 450) 성인 환자를 대상으로 여러 용량의 HUMIRA의 안전성 및 유효성을 평가했습니다. 아미노살리실산염, 코르티코스테로이드 및/또는 면역조절제의 병용 안정 용량이 허용되었으며 환자의 79%가 이러한 약물 중 하나 이상을 계속 투여받았습니다.
두 연구에서 임상적 관해(CDAI < 150으로 정의됨) 유도를 평가했습니다. CD-I 연구에서 299명의 TNF 차단제 순수 치료 환자를 네 가지 치료군 중 하나에 무작위로 배정했습니다. 위약군은 0주차와 2주차에 위약을 투여받았고, 160/80군은 0주차에 160mg의 HUMIRA를, 2주차에 80mg을 투여받았으며, 80/40군은 0주차에 80mg을, 2주차에 40mg을 투여받았고, 40/20군은 0주차에 40mg을, 2주차에 20mg을 투여받았습니다. 4주차에 임상 결과를 평가했습니다.
두 번째 유도 연구인 CD-II 연구에서 이전에 인플릭시맙 치료에 반응이 없거나 내약성이 없는 325명의 환자를 0주차에 160mg의 HUMIRA를, 2주차에 80mg을 투여받거나 0주차와 2주차에 위약을 투여받도록 무작위로 배정했습니다. 4주차에 임상 결과를 평가했습니다.
CD-III 연구에서 임상적 관해 유지를 평가했습니다. 이 연구에서 활동성 질환이 있는 854명의 환자가 0주차에 80mg, 2주차에 40mg의 HUMIRA를 공개 라벨로 투여받았습니다. 그런 다음 4주차에 환자를 격주로 40mg의 HUMIRA를 투여받거나, 매주 40mg의 HUMIRA를 투여받거나, 위약을 투여받도록 무작위로 배정했습니다. 총 연구 기간은 56주였습니다. 4주차에 임상적 반응(CDAI에서 ≥70 감소)을 보인 환자는 4주차에 임상적 반응을 보이지 않은 환자와 별도로 계층화하여 분석했습니다.
임상적 관해 유도
환자가 TNF 차단제 순수 치료 환자인지(CD-I), 인플릭시맙에 반응이 없거나 내약성이 없는지(CD-II)에 관계없이 160/80mg의 HUMIRA로 치료받은 환자의 비율이 4주차에 위약보다 임상적 관해 유도에 도달한 비율이 더 높았습니다(표 12 참조).
| CD-I | CD-II | |||
| 위약 N=74 |
HUMIRA 160/80mg N=76 |
위약 N=166 |
HUMIRA 160/80mg N=159 |
|
| 4주차 | ||||
| 임상적 관해 | 12% | 36%* | 7% | 21%* |
| 임상적 반응 | 34% | 58%** | 34% | 52%** |
| 임상적 관해는 CDAI 점수 < 150이고, 임상적 반응은 CDAI가 최소 70점 감소한 것입니다. * HUMIRA vs. 위약 비율의 쌍별 비교에 대한 p<0.001 ** HUMIRA vs. 위약 비율의 쌍별 비교에 대한 p<0.01 |
||||
임상적 관해 유지
CD-III 연구의 4주차에 환자의 58%(499/854)가 임상 반응을 보였으며 1차 분석에서 평가되었습니다. 26주차와 56주차에 4주차에 임상 반응을 보인 환자 중 위약 유지 그룹의 환자에 비해 HUMIRA 40mg 격주 유지 그룹에서 임상적 관해에 도달한 환자의 비율이 더 높았습니다(표 13 참조). 매주 HUMIRA 치료를 받은 그룹은 격주로 HUMIRA를 투여받은 그룹에 비해 유의하게 높은 관해율을 보이지 않았습니다.
| 위약 | 40mg HUMIRA 격주 투여 |
|
| N=170 | N=172 | |
| 26주차 | ||
| 임상적 관해 | 17% | 40%* |
| 임상 반응 | 28% | 54%* |
| 56주차 | ||
| 임상적 관해 | 12% | 36%* |
| 임상 반응 | 18% | 43%* |
| 임상적 관해는 CDAI 점수 < 150입니다. 임상 반응은 CDAI가 최소 70점 감소한 것입니다. *p<0.001(HUMIRA 대 위약 비율의 쌍별 비교) |
||
4주차에 반응을 보인 환자 중 연구 기간 동안 관해에 도달한 환자의 경우, HUMIRA 격주 투여군이 위약 유지군보다 관해 유지 기간이 더 길었습니다. 12주까지 반응을 보이지 않은 환자의 경우, 12주를 초과하여 치료를 계속해도 유의미하게 더 많은 반응이 나타나지 않았습니다.
14.6 소아 크론병
중등도에서 중증의 활동성 크론병(소아 크론병 활성 지수[PCDAI] 점수 > 30으로 정의)이 있는 192명의 소아 환자(6~17세)를 대상으로 2가지 용량 농도의 HUMIRA에 대한 무작위, 이중맹검, 52주 임상 연구(연구 PCD-I)를 수행했습니다. 등록된 환자는 지난 2년 동안 코르티코스테로이드 또는 면역조절제(예: 아자티오프린, 6-머캅토퓨린 또는 메토트렉세이트)에 대한 반응이 불충분했습니다. 이전에 TNF 차단제를 투여받은 환자는 해당 TNF 차단제에 대한 반응 손실 또는 불내성이 있는 경우 등록할 수 있었습니다.
환자는 체중(≥40kg 및 <40kg)에 따라 용량을 정하여 공개 유도 요법을 받았습니다. 체중이 ≥40kg인 환자는 160mg(0주차) 및 80mg(2주차)을 투여받았습니다. 체중이 <40kg인 환자는 80mg(0주차) 및 40mg(2주차)을 투여받았습니다. 4주차에 각 체중 범주(≥40kg 및 <40kg) 내의 환자를 두 가지 유지 용량 요법(고용량 및 저용량) 중 하나에 1:1로 무작위 배정했습니다. 고용량은 체중이 ≥40kg인 환자의 경우 40mg 격주였고 체중이 <40kg인 환자의 경우 20mg 격주였습니다. 저용량은 체중이 ≥40kg인 환자의 경우 20mg 격주였고 체중이 <40kg인 환자의 경우 10mg 격주였습니다.
연구 전반에 걸쳐 안정적인 용량의 코르티코스테로이드(프레드니손 용량 ≤40mg/일 또는 이에 상응하는 용량) 및 면역조절제(아자티오프린, 6-머캅토퓨린 또는 메토트렉세이트)의 병용 투여가 허용되었습니다.
12주차에 질병 재발(4주차부터 PCDAI가 ≥ 15 증가하고 절대 PCDAI > 30)을 경험했거나 무반응자(최소 2주 간격으로 2회 연속 방문 시 기준치보다 PCDAI가 ≥ 15 감소하지 않음)인 환자는 용량을 증량할 수 있었습니다(즉, 맹검 격주 투여에서 맹검 매주 투여로 전환). 용량을 증량한 환자는 치료 실패로 간주했습니다.
기준치에서 환자의 38%가 코르티코스테로이드를 투여받고 있었고 62%가 면역조절제를 투여받고 있었습니다. 환자의 44%가 이전에 TNF 차단제에 대한 반응 손실 또는 불내성을 경험했습니다. 평균 기준치 PCDAI 점수는 40점이었습니다.
총 192명의 환자 중 188명이 4주 유도 기간을 완료했고, 152명이 26주 치료를 완료했으며, 124명이 52주 치료를 완료했습니다. 저용량 유지 용량군의 51%(48/95)와 고용량 유지 용량군의 38%(35/93)에서 용량을 증량했습니다.
4주차에 환자의 28%(52/188)가 임상적 관해(PCDAI ≤ 10으로 정의)에 도달했습니다.
26주차와 52주차에 임상적 관해(PCDAI ≤ 10으로 정의) 및 임상적 반응(기준치보다 PCDAI가 최소 15점 감소한 것으로 정의)에 있는 환자의 비율을 평가했습니다.
26주차와 52주차 모두에서 임상적 관해 및 임상적 반응을 보인 환자의 비율은 저용량군에 비해 고용량군에서 수치적으로 더 높았습니다(표 14). 권장 유지 용량 요법은 체중이 < 40kg인 환자의 경우 20mg 격주이고 체중이 ≥ 40kg인 환자의 경우 40mg 격주입니다. 매주 투여는 권장 유지 용량 요법이 아닙니다[용량 및 투여 (2.3)] 참조].
| 저용량 유지 용량† (20mg 또는 10mg 격주) N = 95 |
고용량 유지 용량# (40mg 또는 20mg 격주) N = 93 |
|
| 26주차 | ||
| 임상적 관해‡ | 28% | 39% |
| 임상적 반응§ | 48% | 59% |
| 52주차 | ||
| 임상적 관해‡ | 23% | 33% |
| 임상적 반응§ | 28% | 42% |
| †저용량 유지 용량은 체중이 ≥ 40kg인 환자의 경우 20mg 격주였고 체중이 < 40kg인 환자의 경우 10mg 격주였습니다. #고용량 유지 용량은 체중이 ≥ 40kg인 환자의 경우 40mg 격주였고 체중이 < 40kg인 환자의 경우 20mg 격주였습니다. ‡임상적 관해는 PCDAI ≤ 10으로 정의됩니다. §임상적 반응은 기준치보다 PCDAI가 최소 15점 감소한 것으로 정의됩니다. |
||
14.7 성인 궤양성 대장염
HUMIRA의 안전성 및 유효성은 코르티코스테로이드, 아자티오프린 또는 6-MP와 같은 면역억제제를 병용 또는 사전에 투여했음에도 불구하고 중등도에서 중증의 활동성 궤양성 대장염(Mayo 점수 6~12점, 내시경 검사 점수 2~3점, 0~3점 척도 기준)을 앓고 있는 성인 환자를 대상으로 무작위 배정, 이중맹검, 위약 대조 임상 시험 2건(UC-I 연구 및 UC-II 연구)에서 평가되었습니다. 두 연구 모두 TNF 차단제를 투여한 적이 없는 환자를 등록했지만 UC-II 연구에서는 TNF 차단제에 대한 반응이 없거나 내약성이 없는 환자도 등록할 수 있었습니다. UC-II 연구에 등록된 환자의 40%는 이전에 다른 TNF 차단제를 사용한 적이 있었습니다.
아미노살리실산염 및 면역억제제의 병용 안정 용량이 허용되었습니다. UC-I 연구 및 II 연구에서 환자들은 기준 시점에 아미노살리실산염(69%), 코르티코스테로이드(59%) 및/또는 아자티오프린 또는 6-MP(37%)를 투여받고 있었습니다. 두 연구 모두에서 환자의 92%가 이러한 약물 중 하나 이상을 투여받았습니다.
8주차에 임상적 관해(Mayo 점수 ≤ 2, 개별 점수 > 1 없음으로 정의) 유도를 두 연구 모두에서 평가했습니다. 52주차에 임상적 관해 및 지속적인 임상적 관해(8주차와 52주차 모두에서 임상적 관해로 정의)를 UC-II 연구에서 평가했습니다.
UC-I 연구에서 TNF 차단제를 투여한 적이 없는 환자 390명을 1차 유효성 분석을 위해 세 가지 치료군 중 하나에 무작위 배정했습니다. 위약군은 0, 2, 4, 6주차에 위약을 투여받았습니다. 160/80군은 0주차에 HUMIRA 160mg을, 2주차에 80mg을 투여받았고 80/40군은 0주차에 HUMIRA 80mg을, 2주차에 40mg을 투여받았습니다. 2주차 이후 두 HUMIRA 치료군의 환자는 격주로 40mg을 투여받았습니다.
UC-II 연구에서 518명의 환자를 0주차에 HUMIRA 160mg, 2주차에 80mg, 4주차부터 50주차까지 격주로 40mg을 투여받는 군 또는 0주차부터 50주차까지 격주로 위약을 투여받는 군에 무작위 배정했습니다. 8주차부터 코르티코스테로이드 감량이 허용되었습니다.
UC-I 연구 및 UC-II 연구 모두에서 위약으로 치료받은 환자에 비해 HUMIRA 160/80mg으로 치료받은 환자의 비율이 임상적 관해 유도에 도달한 비율이 더 높았습니다. UC-II 연구에서 위약으로 치료받은 환자에 비해 HUMIRA 160/80mg으로 치료받은 환자의 비율이 지속적인 임상적 관해(8주차와 52주차 모두에서 임상적 관해)에 도달한 비율이 더 높았습니다(표 15).
| UC-I 연구 | UC-II 연구 | |||||
| 위약 N=130 |
HUMIRA 160/80mg N=130 |
치료 차이 (95% CI) |
위약 N=246 |
HUMIRA 160/80mg N=248 |
치료 차이 (95% CI) |
|
| 임상적 관해 유도(8주차에 임상적 관해) | 9.2% | 18.5% | 9.3%* (0.9%, 17.6%) |
9.3% | 16.5% | 7.2%* (1.2%, 12.9%) |
| 지속적인 임상적 관해(8주차와 52주차 모두에서 임상적 관해) | 해당 없음 | 해당 없음 | 해당 없음 | 4.1% | 8.5% | 4.4%* (0.1%, 8.6%) |
| 임상적 관해는 Mayo 점수 ≤ 2, 개별 점수 > 1 없음으로 정의됩니다. CI=신뢰 구간 * HUMIRA vs. 위약 비율의 쌍별 비교에 대한 p<0.05 |
||||||
UC-I 연구에서 8주차에 HUMIRA 80/40mg 투여군과 위약 투여군 간에 임상적 관해에서 통계적으로 유의미한 차이가 관찰되지 않았습니다.
UC-II 연구에서 52주차에 HUMIRA 투여군의 17.3%(43/248)가 위약 투여군의 8.5%(21/246)와 비교하여 임상적 관해에 도달했습니다(치료 차이: 8.8%, 95% 신뢰 구간(CI): [2.8%, 14.5%], p<0.05).
UC-II 연구에서 이전에 TNF 차단제를 사용한 적이 있는 환자 하위군에서 임상적 관해 유도에 대한 치료 차이는 전체 연구 모집단에서 관찰된 것보다 더 낮게 나타났으며, 지속적인 임상적 관해 및 52주차에 임상적 관해에 대한 치료 차이는 전체 연구 모집단에서 관찰된 것과 유사하게 나타났습니다. 이전에 TNF 차단제를 사용한 적이 있는 환자 하위군은 HUMIRA 투여군의 9%(9/98)가 위약 투여군의 7%(7/101)와 비교하여 임상적 관해 유도에 도달했으며, HUMIRA 투여군의 5%(5/98)가 위약 투여군의 1%(1/101)와 비교하여 지속적인 임상적 관해에 도달했습니다. 이전에 TNF 차단제를 사용한 적이 있는 환자 하위군에서 52주차에 HUMIRA 투여군의 10%(10/98)가 위약 투여군의 3%(3/101)와 비교하여 임상적 관해에 도달했습니다.
14.8 소아 궤양성 대장염
HUMIRA의 안전성 및 유효성은 중등도 내지 중증의 활동성 궤양성 대장염(내시경 검사 하위 점수 2~3점으로 Mayo 점수 6~12점이며, 중앙 판독 내시경 검사로 확진)이 있고 코르티코스테로이드 및/또는 면역조절제(즉, 아자티오프린, 6-머캅토퓨린 또는 메토트렉세이트)로 치료에 불충분한 반응을 보이거나 내약성이 없는 5세~17세 소아 환자 93명을 대상으로 한 다기관, 무작례화, 이중맹검 시험(연구 PUC-I, NCT02065557)에서 평가되었습니다. 연구에 참여한 93명의 환자 중 15명(16%)은 이전에 TNF 차단제를 사용한 경험이 있었습니다. 등록 시 코르티코스테로이드를 투여받은 환자는 4주 후에 코르티코스테로이드 치료를 점감할 수 있었습니다.
77명의 환자를 처음에 3:2 비율로 무작위 배정하여 두 가지 HUMIRA 용량 중 하나로 이중맹검 치료를 받도록 했습니다. 두 용량군의 환자는 0주차에 2.4mg/kg(최대 160mg), 2주차에 1.2mg/kg(최대 80mg), 4주차와 6주차에 0.6mg/kg(최대 40mg)을 투여받았습니다. 고용량군은 또한 1주차에 2.4mg/kg(최대 160mg)의 추가 용량을 투여받았습니다. 연구 설계가 수정된 후 16명의 환자가 추가로 등록되어 고용량의 HUMIRA를 사용한 개방형 라벨 치료를 받았습니다.
8주차에 부분 Mayo 점수(PMS, 내시경 검사 구성 요소가 없는 Mayo 점수의 하위 집합이며 PMS 감소 ≥ 2점 및 기준치에서 ≥ 30% 감소로 정의됨)에 따라 임상적 반응을 보인 62명의 환자를 무작위 배정하여 HUMIRA 0.6mg/kg(최대 40mg)을 격주로 투여(저용량군)하거나 0.6mg/kg(최대 40mg)을 매주 투여(고용량군)하는 이중맹검 치료를 받도록 했습니다. 연구 설계가 수정되기 전에 PMS에 따라 임상적 반응을 보인 12명의 환자가 추가로 무작위 배정되어 위약을 투여받았습니다.
52주 PUC-I 임상시험에서 연구된 고용량과 권장 HUMIRA 용량 간에 임상적으로 유의미한 유효성 차이는 예상되지 않습니다 [용법 및 용량 (2.4), 임상 약리학 (12.2)]를 참조하십시오].
12주차 또는 그 이후에 질병 악화 기준을 충족한 환자는 무작위 배정되어 2.4mg/kg(최대 160mg)의 재유도 용량 또는 0.6mg/kg(최대 40mg)의 용량을 투여받은 후 8주차에 무작위 배정된 용량을 계속 투여받았습니다.
이 연구의 공동 1차 평가변수는 8주차에 PMS에 따른 임상적 관해(PMS ≤ 2점 및 개별 하위 점수 > 1점 없음으로 정의됨)였고, 8주차에 PMS에 따라 임상적 반응을 달성한 환자에서 52주차에 Mayo 점수에 따른 임상적 관해(Mayo 점수 ≤ 2점 및 개별 하위 점수 > 1점 없음으로 정의됨)였습니다. 2차 평가변수에는 8주차에 PMS에 반응을 보인 환자에서 52주차에 Mayo 점수 반응(Mayo 점수 감소 ≥ 3점 및 기준치에서 ≥ 30% 감소로 정의됨), 8주차에 PMS에 반응을 보인 환자에서 52주차에 내시경 검사 개선(Mayo 내시경 검사 하위 점수 ≤ 1점으로 정의됨), 8주차에 PMS에 따라 관해를 보인 환자에서 52주차에 Mayo 점수 관해가 포함되었습니다.
8주차 결과
8주차에 고용량군(개방형 라벨 고용량을 투여받은 16명의 환자 제외)의 60%[28/47, 95% 신뢰 구간(CI): (44%, 74%)]와 저용량군의 43%[13/30, 95% CI: (25%, 63%)]에서 PMS 관해에 도달했습니다. 고용량군의 결과는 권장 용량으로 예상되는 결과를 나타냅니다 [용법 및 용량 (2.4), 임상 약리학 (12.2)]를 참조하십시오].
52주차 결과
52주차에 8주차~52주차 사이에 이중맹검 위약, HUMIRA 0.6mg/kg(최대 40mg) 격주 투여 또는 HUMIRA 0.6mg/kg(최대 40mg) 매주 투여를 받은 환자 모집단에서 평가변수를 평가했습니다(표 16).
|
|
위약a
n/N (%), 95% CI |
HUMIRA 최대 40mg(0.6mg/kg) 격주 투여b n/N (%), 95% CI |
HUMIRA 최대 40mg(0.6mg/kg) 매주 투여c n/N (%), 95% CI |
| 8주차 PMS 반응자의 임상적 관해 | 4/12 (33%) (10%, 65%) |
9/31 (29%) (14%, 48%) |
14/31 (45%) (27%, 64%) |
| 8주차 PMS 반응자의 임상적 반응 | 4/12 (33%) (10%, 65%) |
19/31 (61%) (42%, 78%) |
21/31 (68%) (49%, 83%) |
| 8주차 PMS 반응자의 내시경적 개선 | 4/12 (33%) (10%, 65%) |
12/31 (39%) (22%, 58%) |
16/31 (52%) (33%, 70%) |
| 8주차 PMS 관해자의 임상적 관해 | 3/8 (38%) (9%, 76%) |
9/21 (43%) (22%, 66%) |
10/22 (45%) (24%, 68%) |
| CI=신뢰 구간 a 8주차에 PMS 기준 임상적 반응을 보인 12명의 환자가 위약을 투여받도록 무작위 배정되었습니다. 표본 크기가 작기 때문에 위약 데이터 해석에는 제한이 있습니다. b 52주 PUC-I 임상시험에서 연구된 격주 투여량은 권장 HUMIRA 투여량보다 낮습니다 [2.4] 투여 방법 및 용량 참조]. c 52주 PUC-I 임상시험에서 투여된 연구된 고용량과 권장 HUMIRA 용량 간에 효능 면에서 임상적으로 관련된 차이는 예상되지 않습니다. 참고: 52주차에 값이 누락되었거나 질병 재발로 인해 재유도 또는 유지 치료를 받도록 무작위 배정된 환자는 52주차 평가변수에 대한 무반응자로 간주되었습니다. |
|||
14.9 판상 건선
HUMIRA의 안전성 및 효능은 전신 요법 또는 광선 요법의 대상이었던 중등도에서 중증의 만성 판상 건선(Ps) 성인 피험자 1696명을 대상으로 한 무작위배정, 이중맹검, 위약 대조 연구에서 평가되었습니다.
Ps-I 연구에서는 3회의 치료 기간 동안 체표면적(BSA) 침범 ≥10%, 의사의 전체 평가(PGA)에서 최소 중등도의 질병 중증도, 건선 부위 및 중증도 지수(PASI) ≥12인 만성 Ps 환자 1212명을 평가했습니다. A 기간 동안 피험자들은 0주차에 80mg의 초기 용량, 그 후 1주차부터 격주로 40mg의 용량으로 위약 또는 HUMIRA를 투여받았습니다. 16주 치료 후, 16주차에 최소 PASI 75 반응(기준점 대비 PASI 점수가 최소 75% 개선된 것으로 정의됨)을 달성한 피험자들은 B 기간에 들어가 40mg의 HUMIRA를 격주로 공개 라벨로 투여받았습니다. 17주간의 공개 라벨 치료 후, 33주차에 최소 PASI 75 반응을 유지했고 A 기간에 활성 치료를 받도록 처음 무작위 배정되었던 피험자들은 19주 동안 40mg의 HUMIRA를 격주로 또는 위약을 투여받도록 C 기간에 다시 무작위 배정되었습니다. 모든 치료군에서 평균 기준선 PASI 점수는 19였고 기준선 PGA 점수는 “중등도”(53%)에서 “중증”(41%) 및 “매우 중증”(6%)까지였습니다.
Ps-II 연구에서는 BSA 침범 ≥10% 및 PASI ≥12인 만성 판상 건선 환자 중 HUMIRA에 무작위 배정된 99명과 위약에 무작위 배정된 48명을 평가했습니다. 피험자들은 16주 동안 위약 또는 0주차에 80mg의 초기 용량, 그 후 1주차부터 격주로 40mg의 HUMIRA를 투여받았습니다. 모든 치료군에서 평균 기준선 PASI 점수는 21이었고 기준선 PGA 점수는 “중등도”(41%)에서 “중증”(51%) 및 “매우 중증”(8%)까지였습니다.
Ps-I 및 II 연구에서는 6점 PGA 척도에서 “깨끗함” 또는 “최소” 질병을 달성한 피험자의 비율과 16주차에 기준점 대비 PASI 점수가 최소 75% 감소(PASI 75)한 피험자의 비율을 평가했습니다(표 17 및 18 참조).
또한 Ps-I 연구에서는 33주차 이후 및 52주차 또는 그 이전에 “깨끗함” 또는 “최소” 질병의 PGA 또는 PASI 75 반응을 유지한 피험자의 비율을 평가했습니다.
| HUMIRA 40mg 격주 투여 | 위약 | |
| N = 814 | N = 398 | |
| PGA: 깨끗함 또는 최소* | 506 (62%) | 17 (4%) |
| PASI 75 | 578 (71%) | 26 (7%) |
| * 깨끗함 = 플라크 상승 없음, 스케일 없음, 색소과다침착 또는 확산성 분홍색 또는 붉은색 변색 있거나 없음 최소 = 정상 피부보다 플라크가 약간 상승했는지 확인할 수 있거나 없음, 표면 건조증과 약간의 흰색 변색 있거나 없음, 붉은색 변색까지 있거나 없음 |
||
| HUMIRA 40 mg 격주 투여 | 위약 | |
| N = 99 | N = 48 | |
| PGA: 깨끗함 또는 최소* | 70 (71%) | 5 (10%) |
| PASI 75 | 77 (78%) | 9 (19%) |
| * 깨끗함 = 플라크 융기 없음, 스케일링 없음, 과색소침착 또는 분홍색이나 붉은색으로 확산됨 최소 = 정상 피부보다 플라크가 약간 융기되었는지 확인 가능하지만 구분하기 어려움, 표면 건조증과 약간의 흰색 변색, 붉은색 변색까지 나타날 수 있음 |
||
또한, Ps-I 연구에서 33주차에 PASI 75를 유지한 HUMIRA 투여 대상자를 HUMIRA(N = 250) 또는 위약(N = 240)으로 무작위 재배정했습니다. 52주 동안 HUMIRA로 치료한 결과, “깨끗함” 또는 “최소” 질환의 PGA 유지(68% 대 28%) 또는 PASI 75(79% 대 43%)를 기준으로 위약으로 무작위 재배정된 대상자에 비해 HUMIRA를 투여한 대상자에서 효능이 더 오래 유지되었습니다.
총 347명의 안정적인 반응자가 공개 연장 연구에서 중단 및 재투여 평가에 참여했습니다. 재발까지의 중간 시간(PGA “중등도” 이하로 감소)은 약 5개월이었습니다. 중단 기간 동안 농포성 또는 홍피증성 건선으로 전환된 대상자는 없었습니다. 재발한 총 178명의 대상자가 80mg의 HUMIRA로 치료를 다시 시작했고, 1주차부터 격주로 40mg을 투여했습니다. 16주차에 대상자의 69%(123/178)가 PGA “깨끗함” 또는 “최소” 반응을 보였습니다.
무작위맹검 연구(Ps-III 연구)에서 217명의 성인 대상자를 대상으로 HUMIRA와 위약의 효능 및 안전성을 비교했습니다. 연구 대상자는 PGA 척도에서 최소 중등도의 만성 플라크 건선, 5점 손톱 건선에 대한 의사의 전체 평가(PGA-F) 척도에서 최소 중등도의 손톱 침범, 표적 손톱에 대한 수정된 손톱 건선 중증도 지수(mNAPSI) 점수 ≥ 8, 그리고 BSA 침범이 최소 10% 이상이거나 모든 손톱에 대한 총 mNAPSI 점수가 ≥ 20점인 BSA 침범이 최소 5% 이상이어야 했습니다. 대상자는 초기 용량 80mg의 HUMIRA를 투여받은 후 26주 동안 격주로 40mg(초기 용량 투여 1주일 후 시작)을 투여받거나 위약을 투여받았으며, 이후 26주 동안 공개 라벨 HUMIRA 치료를 받았습니다. 이 연구에서는 26주차에 PGA-F 척도에서 최소 2등급 개선과 함께 “깨끗함” 또는 “최소” 평가를 달성한 대상자의 비율과 mNAPSI 점수에서 기준치보다 최소 75% 개선을 달성한 대상자의 비율(mNAPSI 75)을 평가했습니다.
26주차에 위약군보다 HUMIRA군에서 PGA-F 종료점에 도달한 대상자의 비율이 더 높았습니다. 또한, 26주차에 위약군보다 HUMIRA군에서 mNAPSI 75에 도달한 대상자의 비율이 더 높았습니다(표 19 참조).
| 종료점 | HUMIRA 40 mg 격주 투여* N=109 |
위약 N=108 |
| PGA-F: ≥2등급 개선 및 깨끗함 또는 최소 | 49% | 7% |
| mNAPSI 75 | 47% | 3% |
| *대상자는 0주차에 80mg의 HUMIRA를 투여받았고, 1주차부터 격주로 40mg을 투여받았습니다. | ||
손발톱 통증도 평가되었으며, 연구 Ps-III에서 손발톱 통증의 개선이 관찰되었습니다.
14.10 화농성 한선염
두 건의 무작위배정, 이중맹검, 위약대조 연구(연구 HS-I 및 II)에서 중등도에서 중증의 Hurley II기 또는 III기 질환을 앓고 있으며 농양이나 염증성 결절이 3개 이상 있는 성인 화농성 한선염(HS) 환자 633명을 대상으로 HUMIRA의 안전성 및 유효성을 평가했습니다. 두 연구 모두에서 환자들은 0주차에 160mg의 초기 용량, 2주차에 80mg, 4주차부터 11주차까지 매주 40mg의 HUMIRA 또는 위약을 투여받았습니다. 환자들은 매일 국소 소독제로 세척했습니다. 연구 HS-II에서는 병용 경구 항생제 사용이 허용되었습니다.
두 연구 모두 12주차에 화농성 한선염 임상 반응(HiSCR)을 평가했습니다. HiSCR은 기준치 대비 총 농양 및 염증성 결절 수가 50% 이상 감소하고 농양 수가 증가하지 않으며 배농 누공 수가 증가하지 않는 것으로 정의되었습니다(표 18 참조). 11점 척도에서 초기 기준 점수가 3점 이상인 환자에서 HS 관련 피부 통증의 감소를 숫자 등급 척도를 사용하여 평가했습니다.
두 연구 모두에서 위약 치료군보다 HUMIRA 치료군에서 HiSCR에 도달한 환자의 비율이 더 높았습니다(표 20 참조).
| HS 연구 I | HS 연구 II* | |||
| 위약 | Humira 40mg 매주 투여 | 위약 | Humira 40mg 매주 투여 | |
| 화농성 한선염 임상 반응(HiSCR) | N = 154 40 (26%) |
N = 153 64 (42%) |
N=163 45 (28%) |
N=163 96 (59%) |
| *연구 HS-II의 환자 중 19.3%는 연구 기간 동안 기준 경구 항생제 치료를 계속했습니다. | ||||
두 연구 모두 12주차에서 35주차(기간 B)까지 HUMIRA를 투여받은 환자들을 3개의 치료군(매주 HUMIRA 40mg, 격주 HUMIRA 40mg 또는 위약) 중 하나에 무작위로 재배정했습니다. 위약을 무작위로 배정받은 환자들은 매주 HUMIRA 40mg(HS-I 연구) 또는 위약(HS-II 연구)을 투여받도록 배정되었습니다.
기간 B 동안 농양 및 염증성 결절 수의 기준치보다 ≥25% 증가하고 최소 2개 이상의 병변이 있는 것으로 정의된 HS의 재발은 두 연구에서 1차 유효성 평가 시점 이후 HUMIRA 치료를 중단한 100명의 환자 중 22명(22%)에서 나타났습니다.
14.11 성인 포도막염
단독 전방 포도막염 환자를 제외한 비감염성 중간 포도막염, 후방 포도막염 및 전체 포도막염 성인 환자를 대상으로 한 두 건의 무작위배정, 이중맹검, 위약대조 연구(UV I 및 II)에서 HUMIRA의 안전성 및 유효성을 평가했습니다. 환자들은 위약 또는 초기 용량 80mg의 HUMIRA를 투여받은 후 초기 용량 투여 1주일 후부터 격주로 40mg을 투여받았습니다. 두 연구의 1차 유효성 평가변수는 ‘치료 실패까지의 시간’이었습니다.
치료 실패는 새로운 염증성 맥락망막 및/또는 염증성 망막 혈관 병변의 발생, 전방 카메라(AC) 세포 등급 또는 유리체 혼탁(VH) 등급의 증가 또는 최대 교정 시력(BCVA)의 감소로 정의된 다중 구성 요소 결과였습니다.
UV I 연구에서는 코르티코스테로이드(프레드니손 1일 10~60mg 경구 투여)로 치료받고 있는 활동성 포도막염 환자 217명을 평가했습니다. 모든 환자는 연구 시작 시 표준 용량인 프레드니손 60mg/일을 투여받은 후 15주차까지 코르티코스테로이드를 완전히 중단하는 필수적인 감량 일정을 따랐습니다.
UV II 연구에서는 기저 질환을 조절하기 위해 코르티코스테로이드(프레드니손 10~35mg/일 경구 투여)로 치료받고 있는 비활동성 포도막염 환자 226명을 평가했습니다. 그 후 환자들은 19주차까지 코르티코스테로이드를 완전히 중단하는 필수적인 감량 일정을 따랐습니다.
임상 반응
두 연구 결과 모두 위약을 투여받은 환자에 비해 HUMIRA를 투여받은 환자에서 치료 실패 위험이 통계적으로 유의하게 감소한 것으로 나타났습니다. 두 연구 모두에서 1차 평가변수의 모든 구성 요소가 HUMIRA군과 위약군 간의 전반적인 차이에 누적적으로 기여했습니다(표 21).
| UV I | UV II | |||||
| 위약 (N = 107) |
HUMIRA (N = 110) |
HR [95% CI]a |
위약 (N = 111) |
HUMIRA (N = 115) |
HR [95% CI]a |
|
| 실패b n (%) | 84 (78.5) | 60 (54.5) | 0.50 [0.36, 0.70] |
61 (55.0) | 45 (39.1) | 0.57 [0.39, 0.84] |
| 실패까지의 중간 시간(개월) [95% CI] |
3.0 [2.7, 3.7] |
5.6 [3.9, 9.2] |
해당 없음 | 8.3 [4.8, 12.0] |
추정 불가c | 해당 없음 |
| ª 치료를 요인으로 하는 비례 위험 회귀 분석에서 HUMIRA 대 위약의 HR. b UV I 연구에서 6주 이후 또는 UV II 연구에서 2주 이후의 치료 실패는 사건으로 간주되었습니다. 연구를 중단한 환자는 탈락 시점에 중단된 것으로 간주되었습니다. c 추정 불가 = 추정 불가능. 위험에 처한 대상의 절반 미만에서 사건이 발생했습니다. |
||||||
그림 3: 6주(UV I 연구) 또는 2주(UV II 연구) 후 치료 실패까지의 시간을 요약한 카플란-마이어 곡선
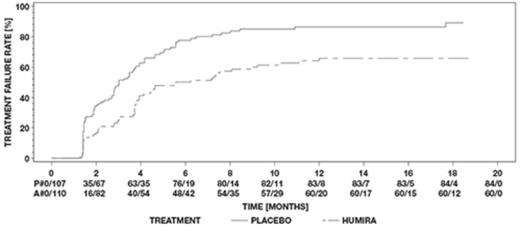
UV I 연구
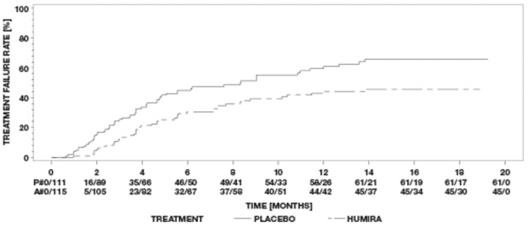
UV II 연구
참고: P# = 위약(사건 수/위험군 수); A# = HUMIRA(사건 수/위험군 수).
14.12 소아 포도막염
활동성 JIA 관련 비감염성 포도막염(PUV-I)이 있는 2~<18세 소아 환자 90명을 대상으로 한 무작위, 이중맹검, 위약 대조 연구에서 HUMIRA의 안전성 및 유효성을 평가했습니다. 환자들은 메토트렉세이트 용량과 함께 격주로 위약 또는 아달리무맙 20mg(30kg 미만) 또는 아달리무맙 40mg(30kg 이상)을 투여받았습니다. 연구 시작 시 병용 코르티코스테로이드 용량이 허용되었으며, 이후 3개월 이내에 국소 코르티코스테로이드를 의무적으로 감량했습니다.
1차 평가변수는 ‘치료 실패까지의 시간’이었습니다. 치료 실패를 결정하는 기준은 안구 염증의 악화 또는 지속적인 비개선 또는 안구 동반 질환의 악화였습니다.
임상 반응
HUMIRA는 위약에 비해 치료 실패 위험을 75% 감소시켰습니다(HR = 0.25 [95% CI: 0.12, 0.49])(표 22).
| 위약 (N=30) |
HUMIRA (N=60) |
HR (95% CI)ª | |
| 실패(n[%]) | 18 (60%) | 16 (26.7%) | 0.25 (0.12, 0.49) |
| 실패까지의 중앙값(주) (95% CI)ᵇ |
24.1 (12.4, 81.0) |
NEᶜ | |
| a 치료를 요인으로 하는 비례 위험 회귀 분석에서 아달리무맙 대 위약의 HR. b 카플란-마이어 곡선을 기반으로 추정. c NE = 추정 불가. 위험군의 절반 미만에서 사건이 발생했습니다. |
|||
그림 4: 치료 실패까지의 시간을 요약한 카플란-마이어 곡선(PUV-I 연구)
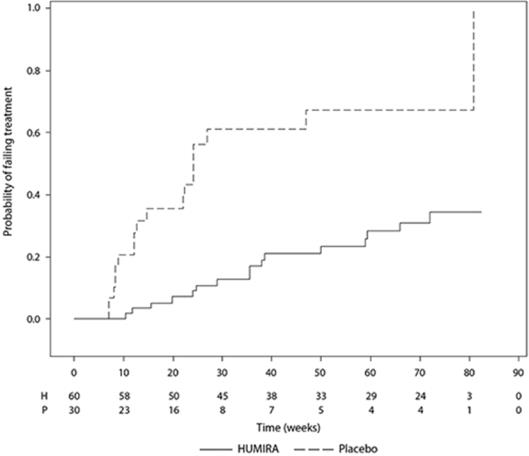
PUV-I 연구
참고: P = 위약(위험군 수); H = HUMIRA(위험군 수).
참고 문헌 15개
- National Cancer Institute. Surveillance, Epidemiology, and End Results Database (SEER) Program. SEER Incidence Crude Rates, 17 Registries, 2000-2007.
16. 공급/보관 및 취급 방법
HUMIRA® (adalimumab)는 피하 주사를 위한 방부제가 없는, 멸균된 투명하고 무색의 용액으로 공급됩니다. 다음과 같은 포장 구성이 가능합니다.
-
HUMIRA 펜 카톤 – 40 mg/0.4 mL
HUMIRA는 두 개의 알코올 준비 용액과 두 개의 투약 트레이가 들어 있는 카톤에 공급됩니다. 각 투약 트레이는 40 mg/0.4 mL의 HUMIRA를 제공하는 고정된 얇은 벽, ½ 인치 바늘이 있는 1 mL 사전 충전 유리 주사기가 들어 있는 단일 투약 펜으로 구성됩니다. 검은색 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다. NDC 번호는 83457-554-02입니다. -
HUMIRA 펜 카톤 – 80 mg/0.8 mL
HUMIRA는 두 개의 알코올 준비 용액과 두 개의 투약 트레이가 들어 있는 카톤에 공급됩니다. 각 투약 트레이는 80 mg/0.8 mL의 HUMIRA를 제공하는 고정된 얇은 벽, ½ 인치 바늘이 있는 1 mL 사전 충전 유리 주사기가 들어 있는 단일 투약 펜으로 구성됩니다. 검은색 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다. NDC 번호는 83457-124-02입니다. -
사전 충전 주사기 카톤 – 40 mg/0.4 mL
HUMIRA는 두 개의 알코올 준비 용액과 두 개의 투약 트레이가 들어 있는 카톤에 공급됩니다. 각 투약 트레이는 40 mg/0.4 mL의 HUMIRA를 제공하는 고정된 얇은 벽, ½ 인치 바늘이 있는 단일 투약, 1 mL 사전 충전 유리 주사기로 구성됩니다. 검은색 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다. NDC 번호는 83457-243-02입니다. -
사전 충전 주사기 카톤 – 20 mg/0.2 mL
HUMIRA는 두 개의 알코올 준비 용액과 두 개의 투약 트레이가 들어 있는 카톤에 공급됩니다. 각 투약 트레이는 20 mg/0.2 mL의 HUMIRA를 제공하는 고정된 얇은 벽, ½ 인치 바늘이 있는 단일 투약, 1 mL 사전 충전 유리 주사기로 구성됩니다. 검은색 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다. NDC 번호는 83457-616-02입니다. -
사전 충전 주사기 카톤 – 10 mg/0.1 mL
HUMIRA는 두 개의 알코올 준비 용액과 두 개의 투약 트레이가 들어 있는 카톤에 공급됩니다. 각 투약 트레이는 10 mg/0.1 mL의 HUMIRA를 제공하는 고정된 얇은 벽, ½ 인치 바늘이 있는 단일 투약, 1 mL 사전 충전 유리 주사기로 구성됩니다. 검은색 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다. NDC 번호는 83457-817-02입니다.
보관 및 안정성
용기의 유효 기간이 지나면 사용하지 마십시오. HUMIRA는 36°F에서 46°F(2°C에서 8°C) 사이의 냉장 보관해야 합니다. 냉동하지 마십시오. 냉동된 경우 해동하더라도 사용하지 마십시오.
빛으로부터 보호하기 위해 투약 전까지는 원래 카톤에 보관하십시오.
필요한 경우, 예를 들어 여행 중에는 HUMIRA를 최대 77°F(25°C)의 실온에 최대 14일 동안 보관할 수 있지만 빛으로부터 보호해야 합니다. HUMIRA는 14일 이내에 사용하지 않으면 폐기해야 합니다. HUMIRA를 냉장고에서 처음 꺼낸 날짜를 카톤과 투약 트레이에 제공된 공간에 기록하십시오.
HUMIRA를 극심한 열이나 추위에 보관하지 마십시오.
17 환자 상담 정보
환자 또는 보호자에게 FDA 승인 환자 라벨(약물 안내 및 사용 지침)을 읽도록 알려주십시오.
감염
환자에게 HUMIRA가 감염과 싸우는 면역 체계의 능력을 떨어뜨릴 수 있다고 알려주십시오. 환자에게 결핵, 침윤성 진균 감염 및 B형 간염 바이러스 감염 재활성화를 포함한 감염 증상이 나타나면 의사에게 연락하는 것이 중요하다는 것을 알려주십시오 [경고 및 주의 사항 (5.1, 5.2, 5.4)].
악성 종양
환자에게 HUMIRA를 투여받는 동안 악성 종양의 위험성에 대해 상담하십시오 [경고 및 주의 사항 (5.2)]
과민 반응
환자에게 심각한 과민 반응 증상이 나타나면 즉시 의료 처치를 받도록 알려주십시오. 라텍스 알레르기가 있는 환자에게 HUMIRA 40 mg/0.8 mL 펜 및 40 mg/0.8 mL, 20 mg/0.4 mL 및 10 mg/0.2 mL 미리 채워진 주사기의 바늘 캡에 천연 고무 라텍스가 포함될 수 있다고 알려주십시오 [경고 및 주의 사항 (5.3), 공급/보관 및 취급 방법 (16)].
기타 의학적 상태
환자에게 울혈성 심부전, 신경 질환, 자가 면역 질환 또는 혈구 감소증과 같은 새로운 의학적 상태 또는 악화되는 의학적 상태의 징후를 보고하도록 알려주십시오. 환자에게 멍, 출혈 또는 지속적인 발열과 같은 혈구 감소증을 시사하는 증상을 보고하도록 알려주십시오 [경고 및 주의 사항 (5.5, 5.6, 5.8, 5.9)].
주사 기술에 대한 지침
환자에게 첫 번째 주사는 자격을 갖춘 의료 전문가의 감독하에 시행되어야 한다고 알려주십시오. 환자 또는 보호자가 HUMIRA를 투여하는 경우, 주사 기술을 지시하고 HUMIRA가 적절하게 투여되도록 피하 주사를 할 수 있는지 평가하십시오 [사용 지침 (사용 지침) 참조].
HUMIRA 펜을 사용할 환자에게 다음과 같이 알려주십시오.
- 자주색 활성화 버튼을 누르면 큰 ‘딸깍’ 소리가 들립니다. 큰 딸깍 소리는 주사의 시작을 의미합니다.
- 약물이 모두 주입될 때까지 HUMIRA 펜을 꽉 쥐고 들어올린 피부에 대고 있어야 합니다. 이는 최대 15초가 걸릴 수 있습니다.
- 노란색 표시가 창에 완전히 나타나고 움직임을 멈추면 주사가 완료된 것입니다.
환자에게 사용한 바늘과 주사기 또는 사용한 펜을 사용 후 즉시 FDA 승인 폐기 용기에 버리도록 지시하십시오. 환자에게 사용한 바늘과 주사기 또는 펜을 가정 쓰레기통에 버리지 말라고 지시하십시오. 환자에게 FDA 승인 폐기 용기가 없는 경우, 내구성이 강한 플라스틱으로 만들어지고, 바늘이 빠져나올 수 없는 꽉 끼는 뚜껑으로 닫을 수 있으며, 사용 중에 똑바로 서고 안정적이며, 누출 방지 기능이 있으며, 용기 내부의 위험한 폐기물을 경고하는 적절한 라벨이 붙어 있는 가정용 용기를 사용할 수 있다고 지시하십시오.
환자에게 폐기 용기가 거의 가득 차면, 폐기 용기를 올바르게 폐기하는 방법에 대한 지역 사회 지침을 따라야 한다고 지시하십시오. 환자에게 사용한 바늘과 주사기를 폐기하는 것에 대한 주 또는 지역 법률이 있을 수 있다고 지시하십시오. 환자에게 안전한 폐기 방법에 대한 자세한 정보와 거주하는 주의 폐기 방법에 대한 구체적인 정보를 얻으려면 FDA 웹사이트 http://www.fda.gov/safesharpsdisposal을 참조하도록 지시하십시오.
환자에게 지역 사회 지침에서 허용하지 않는 한 사용한 폐기 용기를 가정 쓰레기통에 버리지 말라고 지시하십시오. 환자에게 사용한 폐기 용기를 재활용하지 말라고 지시하십시오.
AbbVie Inc.
North Chicago, IL 60064, U.S.A.
US License Number 1889
20082819 11/2023
Manufactured for:
Cordavis Trading Limited
Dublin, Ireland
투약 안내
| 투약 안내 HUMIRA® (Hu-MARE-ah) (adalimumab) 주사제, 피하 주사용 |
||
| HUMIRA를 복용하기 전과 재충전할 때마다 HUMIRA와 함께 제공되는 투약 안내를 읽으십시오. 새로운 정보가 있을 수 있습니다. 이 투약 안내는 의사와 귀하의 질병이나 치료에 대해 상담하는 것을 대신할 수 없습니다. | ||
| HUMIRA에 대해 알아야 할 가장 중요한 정보는 무엇입니까? HUMIRA는 면역 체계에 영향을 미치는 약물입니다. HUMIRA는 면역 체계가 감염과 싸우는 능력을 떨어뜨릴 수 있습니다. HUMIRA를 복용하는 사람들에게 심각한 감염이 발생했습니다. 이러한 심각한 감염에는 결핵(TB)과 바이러스, 곰팡이 또는 박테리아로 인한 감염이 포함되며, 이는 신체 전체로 퍼질 수 있습니다. 일부 사람들은 이러한 감염으로 사망했습니다.
의사가 허용하지 않는 한 감염이 있는 경우 HUMIRA를 복용해서는 안 됩니다.
|
||
|
|
|
HUMIRA를 시작한 후 감염이나 감염 징후가 나타나면 즉시 의사에게 연락하십시오. |
||
암
|
||
| HUMIRA는 무엇입니까? HUMIRA는 종양괴사인자(TNF) 억제제라고 하는 약물입니다. HUMIRA는 다음과 같은 경우에 사용됩니다.
|
||
HUMIRA가 귀하에게 적합하지 않을 수 있습니다. HUMIRA를 복용하기 전에 다음과 같은 경우를 포함하여 모든 의학적 상태를 의사에게 알리십시오.
- 감염이 있는 경우. “HUMIRA에 대해 알아야 할 가장 중요한 정보는 무엇입니까?”를 참조하십시오.
- 암이 있거나 있었던 경우.
- 마비 또는 저림이 있거나 다발성 경화증 또는 길랭-바레 증후군과 같은 신경계 질환이 있는 경우.
- 심부전이 있거나 있었던 경우.
- 최근에 백신을 접종받았거나 접종받을 예정인 경우. HUMIRA를 사용하는 동안 생백신을 제외한 백신을 접종받을 수 있습니다. 어린이는 HUMIRA를 복용하기 전에 모든 백신을 접종해야 합니다.
- 고무 또는 라텍스 알레르기가 있는 경우. 고무 또는 라텍스에 대한 알레르기가 있는 경우 의사에게 알리십시오.
- HUMIRA Pen 40 mg/0.8 mL, HUMIRA 40 mg/0.8 mL 사전 충전 주사기, HUMIRA 20 mg/0.4 mL 사전 충전 주사기 및 HUMIRA 10 mg/0.2 mL 사전 충전 주사기의 바늘 덮개에는 천연 고무 또는 라텍스가 포함될 수 있습니다.
- HUMIRA Pen 80 mg/0.8 mL, HUMIRA 80 mg/0.8 mL 사전 충전 주사기, HUMIRA Pen 40 mg/0.4 mL, HUMIRA 40 mg/0.4 mL 사전 충전 주사기, HUMIRA 20 mg/0.2 mL 사전 충전 주사기, HUMIRA 10 mg/0.1 mL 사전 충전 주사기의 검은색 바늘 덮개와 HUMIRA 기관 사용 바이알의 바이알 마개는 천연 고무 또는 라텍스로 만들어지지 않았습니다.
- HUMIRA 또는 HUMIRA의 성분에 알레르기가 있는 경우. HUMIRA의 성분 목록은 이 약물 안내서의 끝부분을 참조하십시오.
- 임신 중이거나 임신을 계획하고 있거나 모유 수유 중이거나 모유 수유를 계획하고 있는 경우. 귀하와 의사는 임신 중이거나 모유 수유 중에 HUMIRA를 복용해야 하는지 결정해야 합니다.
- 아기가 있고 임신 중에 HUMIRA를 사용한 경우. 아기가 백신을 접종받기 전에 아기의 의사에게 알리십시오.
처방약과 일반 의약품, 비타민 및 허브 보충제를 포함하여 복용하는 모든 약물에 대해 의사에게 알리십시오.
특히 다음을 사용하는 경우 의사에게 알리십시오.
- ORENCIA (abatacept), KINERET (anakinra), REMICADE (infliximab), ENBREL (etanercept), CIMZIA (certolizumab pegol) 또는 SIMPONI (golimumab)는 이러한 약물 중 하나를 사용하는 동안 HUMIRA를 사용해서는 안 되기 때문입니다.
- RITUXAN (rituximab). 최근에 RITUXAN (rituximab)을 접종받은 경우 의사가 HUMIRA를 투여하지 않을 수 있습니다.
- IMURAN (azathioprine) 또는 PURINETHOL (6–mercaptopurine, 6-MP).
새로운 약을 복용할 때마다 의사와 약사에게 약물 목록을 보여주십시오.
- HUMIRA는 피부 아래에 주사합니다. 의사가 HUMIRA 주사를 얼마나 자주 맞아야 하는지 알려줄 것입니다. 이는 치료해야 할 상태에 따라 다릅니다. 처방된 것보다 자주 HUMIRA를 주사하지 마십시오.
- HUMIRA를 준비하고 주사하는 올바른 방법에 대한 자세한 내용은 상자 안의 사용 지침을 참조하십시오.
- HUMIRA를 직접 주사하기 전에 주사 방법을 보여주었는지 확인하십시오. 주사 방법에 대한 질문이 있으면 의사 또는 1-800-4HUMIRA (1-800-448-6472)로 전화하십시오. HUMIRA를 준비하고 주사하는 방법을 보여준 후에는 아는 사람이 주사를 도울 수도 있습니다.
- 하지 마십시오 주사 방법을 보여주기 전에 HUMIRA를 직접 주사하십시오. 의사가 귀하 또는 보호자가 집에서 HUMIRA를 주사할 수 있다고 판단한 경우 HUMIRA를 준비하고 주사하는 올바른 방법에 대한 교육을 받아야 합니다.
- 의사가 허용하지 않는 한 HUMIRA 복용량을 건너뛰지 마십시오. HUMIRA 복용을 잊은 경우 기억하는 대로 주사하십시오. 그런 다음 다음 복용량을 정기적으로 예정된 시간에 복용하십시오. 이렇게 하면 일정을 다시 시작할 수 있습니다. HUMIRA를 언제 주사해야 할지 확실하지 않으면 의사 또는 약사에게 전화하십시오.
- 처방된 것보다 더 많은 HUMIRA를 복용한 경우 의사에게 전화하십시오.
HUMIRA는 다음을 포함한 심각한 부작용을 유발할 수 있습니다.
“HUMIRA에 대해 알아야 할 가장 중요한 정보는 무엇입니까?”를 참조하십시오.
-
심각한 감염.
의사가 결핵 여부를 검사하고 결핵 검사를 실시할 것입니다. 의사가 결핵 위험이 있다고 판단하는 경우 HUMIRA 치료를 시작하기 전과 HUMIRA 치료 중에 결핵 약물로 치료를 받을 수 있습니다. 결핵 검사 결과가 음성이더라도 의사는 HUMIRA를 복용하는 동안 결핵 감염 여부를 주의 깊게 모니터링해야 합니다. HUMIRA를 복용하기 전에 결핵 피부 반응 검사 결과가 음성이었던 사람들에게 활동성 결핵이 발생했습니다. HUMIRA를 복용하는 동안 또는 복용 후에 다음과 같은 증상이 나타나면 의사에게 알리십시오.
- 사라지지 않는 기침
- 저체온증
- 체중 감소
- 체지방과 근육 손실(소모)
-
혈액 내에 바이러스를 보유한 사람의 B형 간염 감염.
B형 간염 바이러스(간에 영향을 미치는 바이러스)의 보균자인 경우 HUMIRA를 사용하는 동안 바이러스가 활성화될 수 있습니다. 의사는 치료를 시작하기 전, HUMIRA를 사용하는 동안, 그리고 HUMIRA 치료를 중단한 후 몇 달 동안 혈액 검사를 실시해야 합니다. 가능한 B형 간염 감염 증상이 나타나면 의사에게 알리십시오.
- 근육통
- 매우 피곤함
- 짙은 소변
- 피부 또는 눈이 노랗게 보임
- 식욕 부진
- 구토
- 진흙색 변
- 발열
- 오한
- 복통
- 피부 발진
- 알레르기 반응. HUMIRA를 사용하는 사람에게 알레르기 반응이 나타날 수 있습니다. 심각한 알레르기 반응 증상이 나타나면 즉시 의사에게 전화하거나 응급 처치를 받으십시오.
- 두드러기
- 얼굴, 눈, 입술 또는 입의 부기
- 호흡 곤란
- Nervous system problems. Signs and symptoms of a nervous system problem include: numbness or tingling, problems with your vision, weakness in your arms or legs, and dizziness.
- Blood problems. Your body may not make enough of the blood cells that help fight infections or help to stop bleeding. Symptoms include a fever that does not go away, bruising or bleeding very easily, or looking very pale.
- New heart failure or worsening of heart failure you already have. Call your doctor right away if you get new worsening symptoms of heart failure while taking HUMIRA, including:
- shortness of breath
- sudden weight gain
- swelling of your ankles or feet
- Immune reactions including a lupus-like syndrome. Symptoms include chest discomfort or pain that does not go away, shortness of breath, joint pain, or a rash on your cheeks or arms that gets worse in the sun. Symptoms may improve when you stop HUMIRA.
- Liver problems. Liver problems can happen in people who use TNF-blocker medicines. These problems can lead to liver failure and death. Call your doctor right away if you have any of these symptoms:
- feel very tired
- skin or eyes look yellow
- poor appetite or vomiting
- pain on the right side of your stomach (abdomen)
- Psoriasis. Some people using HUMIRA had new psoriasis or worsening of psoriasis they already had. Tell your doctor if you develop red scaly patches or raised bumps that are filled with pus. Your doctor may decide to stop your treatment with HUMIRA.
The most common side effects of HUMIRA include:
- injection site reactions: redness, rash, swelling, itching, or bruising. These symptoms usually will go away within a few days. Call your doctor right away if you have pain, redness or swelling around the injection site that does not go away within a few days or gets worse.
- upper respiratory infections (including sinus infections).
- headaches.
- rash.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
- Store HUMIRA in the refrigerator at 36ºF to 46ºF (2ºC to 8ºC). Store HUMIRA in the original carton until use to protect it from light.
- Do not freeze HUMIRA. Do not use HUMIRA if frozen, even if it has been thawed.
- Refrigerated HUMIRA may be used until the expiration date printed on the HUMIRA carton, dose tray, Pen or prefilled syringe. Do not use HUMIRA after the expiration date.
- If needed, for example when you are traveling, you may also store HUMIRA at room temperature up to 77°F (25°C) for up to 14 days. Store HUMIRA in the original carton until use to protect it from light.
- Throw away HUMIRA if it has been kept at room temperature and not been used within 14 days.
- Record the date you first remove HUMIRA from the refrigerator in the spaces provided on the carton and dose tray.
- Do not store HUMIRA in extreme heat or cold.
- Do not use a Pen or prefilled syringe if the liquid is cloudy, discolored, or has flakes or particles in it.
- Do not drop or crush HUMIRA. The prefilled syringe is glass.
Keep HUMIRA, injection supplies, and all other medicines out of the reach of children.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use HUMIRA for a condition for which it was not prescribed. Do not give HUMIRA to other people, even if they have the same condition. It may harm them.
This Medication Guide summarizes the most important information about HUMIRA. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about HUMIRA that is written for health professionals.
Active ingredient: adalimumab
HUMIRA Pen 40 mg/0.8 mL, HUMIRA 40 mg/0.8 mL prefilled syringe, HUMIRA 20 mg/0.4 mL prefilled syringe, HUMIRA 10 mg/0.2 mL prefilled syringe, and HUMIRA 40 mg/0.8 mL institutional use vial:
Inactive ingredients: citric acid monohydrate, dibasic sodium phosphate dihydrate, mannitol, monobasic sodium phosphate dihydrate, polysorbate 80, sodium chloride, sodium citrate and Water for Injection. Sodium hydroxide is added as necessary to adjust pH.
HUMIRA Pen 80 mg/0.8 mL, HUMIRA 80 mg/0.8 mL prefilled syringe, HUMIRA Pen 40 mg/0.4 mL, HUMIRA 40 mg/0.4 mL prefilled syringe, HUMIRA 20 mg/0.2 mL prefilled syringe and HUMIRA 10 mg/0.1 mL prefilled syringe:
Inactive ingredients: mannitol, polysorbate 80, and Water for Injection.
Manufactured by: AbbVie Inc., North Chicago, IL 60064, U.S.A.
For more information go to www.HUMIRA.com or you can enroll in a patient support program by calling 1-800-4HUMIRA (1-800-448-6472).
US License Number 1889
Manufactured for: Cordavis Trading Limited, Dublin, Ireland
20082821
사용 지침
| 사용 지침 HUMIRA® (휴미라) (아달리무맙) 40 mg/0.4 mL 1회용 펜 |
|
주사하기 전에: 처음 HUMIRA를 사용하기 전에 의료 서비스 제공자가 사용 방법을 보여주어야 합니다. 도움이 필요하면 의료 서비스 제공자에게 전화하거나 1-800-4HUMIRA (1-800-448-6472)로 전화하십시오. |
|
| HUMIRA 주사 전에 알아야 할 중요 정보 | |
| 다음과 같은 경우 펜을 사용하지 말고 의료 서비스 제공자나 약사에게 전화하십시오. | |
|
|
| 주사 직전까지 캡을 씌워 놓으십시오. HUMIRA는 어떻게 보관해야 합니까? |
|
|
|
|
HUMIRA, 주사 용품 및 기타 모든 의약품을 어린이의 손이 닿지 않는 곳에 보관하십시오. HUMIRA 펜을 사용하기 전에 모든 페이지의 지침을 읽으십시오. |
|
 |
냉장고에서 HUMIRA를 꺼내십시오. HUMIRA를 주사하기 전에 15~30분 동안 실온에 두십시오.
|
 |
펜 라벨의 유효 기간을 확인하십시오. 유효 기간이 지난 경우 펜을 사용하지 마십시오. 다음을 깨끗하고 평평한 표면에 놓으십시오.
손을 씻고 말리십시오. |
 |
주사 부위를 선택하십시오.
알코올 솜으로 주사 부위를 원을 그리며 닦으십시오.
|
 |
회색 캡 #1이 위로 향하도록 펜을 잡으십시오. 창을 확인하십시오.
|
 |
회색 캡 #1을 똑바로 잡아당겨 빼십시오. 캡을 버리십시오.
자주색 캡 #2를 똑바로 잡아당겨 빼십시오. |
 |
주사 부위의 피부를 꼬집어 볼록하게 만든 후 주사가 끝날 때까지 단단히 잡습니다. 흰색 화살표를 주사 부위를 향하게 합니다. 흰색 바늘 덮개를 주사 부위에 직각으로 (90° 각도) 대고 놓습니다. 펜을 검사 창이 보이도록 잡습니다. 주사할 준비가 될 때까지 자주색 활성화 버튼을 누르지 마십시오. |
 |
주사를 시작하기 전에 펜을 주사 부위에 완전히 눌러 붙이는 것이 중요합니다. 계속 눌러서 주사하는 동안 펜이 피부에서 떨어지지 않도록 합니다. 자주색 활성화 버튼을 누르고 천천히 10초 동안 셉니다.
|
 |
주사가 완료되면 펜을 피부에서 천천히 빼냅니다. 흰색 바늘 덮개가 바늘 끝을 덮습니다.
주사 부위에 액체가 몇 방울 이상 떨어지면 도움을 받기 위해 1-800-4HUMIRA (1-800-448-6472)로 전화하십시오.
|
 |
사용한 HUMIRA 펜은 어떻게 폐기해야 합니까?
|
| 펜 캡, 알코올 솜, 솜이나 거즈, 용량 트레이, 포장재는 가정 쓰레기통에 버릴 수 있습니다. | |
| HUMIRA 펜 사용에 대한 질문
의료 서비스 제공자로부터 직접 교육을 받지 못했다면 어떻게 해야 합니까?
주사가 완료되었는지 어떻게 알 수 있습니까?
주사 부위에 액체가 몇 방울 이상 떨어지면 어떻게 해야 합니까?
FDA 승인 날카로운 물건 폐기 용기나 적절한 가정용 용기가 없다면 어떻게 해야 합니까?
|
|
| 항상 펜과 날카로운 물건 폐기 용기를 어린이의 손이 닿지 않는 곳에 보관하십시오. | |
 |
주사 날짜와 장소를 기록해 두십시오. HUMIRA를 언제 복용해야 하는지 기억하는 데 도움이 되도록 미리 달력에 표시하십시오. |
 |
|
| 이 사용 지침은 미국 식품의약국(FDA)의 승인을 받았습니다. | 2023년 11월 개정 |
| AbbVie Inc. North Chicago, IL 60064 U.S.A.에서 제조 미국 라이선스 번호 1889 20082820 제조사: Cordavis Trading Limited, Dublin, Ireland |
|
사용 지침
| 사용 지침 | |
| HUMIRA® (휴미라) (아달리무맙) 80 mg/0.8 mL 함유 포장 1회용 펜 |
|
| 주사하기 전에: 처음 HUMIRA를 사용하기 전에 의료 서비스 제공자가 사용 방법을 보여주어야 합니다. 도움이 필요하면 의료 서비스 제공자에게 전화하거나 1-800-4HUMIRA (1-800-448-6472)로 전화하십시오. |
|
| HUMIRA 주사하기 전에 알아야 할 중요한 정보 | |
| 다음과 같은 경우 펜을 사용하지 말고 의료 서비스 제공자나 약사에게 전화하십시오. | |
|
|
| 주사 직전까지 캡을 씌워 두십시오.
HUMIRA는 어떻게 보관해야 합니까? |
|
|
|
|
HUMIRA, 주사 용품 및 기타 모든 의약품을 어린이의 손이 닿지 않는 곳에 보관하십시오. HUMIRA 펜을 사용하기 전에 모든 페이지의 지침을 읽으십시오. |
|
 |
냉장고에서 HUMIRA를 꺼내십시오. HUMIRA를 주사하기 전에 15~30분 동안 실온에 두십시오.
|
|
펜 라벨의 유효 기간을 확인하십시오. 유효 기간이 지난 경우 펜을 사용하지 마십시오. 다음을 깨끗하고 평평한 표면에 놓으십시오.
손을 씻고 말리십시오. |
|
주사 부위를 선택하십시오.
알코올 솜으로 주사 부위를 원을 그리며 닦으십시오.
|
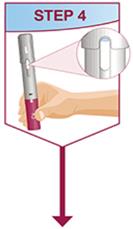 |
회색 캡 #1이 위로 향하도록 펜을 잡으십시오. 창을 확인하십시오.
|
|
회색 캡 #1을 똑바로 잡아당겨 빼십시오. 캡을 버리십시오.
자주색 캡 #2를 똑바로 잡아당겨 빼십시오. |
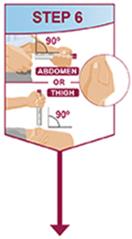 |
주사 부위의 피부를 잡아당겨 돌출되도록 하고 주사가 완료될 때까지 단단히 잡습니다. 흰색 화살표를 주사 부위를 향하게 합니다. 흰색 바늘 덮개를 주사 부위에 직각으로 (90° 각도) 놓습니다. 펜을 검사 창이 보이도록 잡습니다. 주사할 준비가 될 때까지 자주색 활성화 버튼을 누르지 마십시오. |
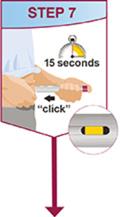 |
주사를 시작하기 전에 펜을 주사 부위에 단단히 눌러 붙이는 것이 중요합니다. 자주색 활성화 버튼을 누르고 15초 동안 천천히 셉니다.
|
|
주사가 완료되면 펜을 피부에서 천천히 빼냅니다. 흰색 바늘 덮개가 바늘 끝을 덮습니다.
주사 부위에 액체가 몇 방울 이상 떨어지면 도움을 받기 위해 1-800-4HUMIRA (1-800-448-6472)로 전화하십시오.
|
|
사용한 HUMIRA 펜은 어떻게 폐기해야 합니까?
|
| 펜 캡, 알코올 솜, 솜이나 거즈, 용량 트레이, 포장은 가정 쓰레기통에 버릴 수 있습니다. | |
| HUMIRA 펜 사용에 대한 질문
의료 서비스 제공자로부터 대면 교육을 받지 못했다면 어떻게 해야 합니까?
주사가 완료되었는지 어떻게 알 수 있습니까?
주사 부위에 액체가 몇 방울 이상 떨어지면 어떻게 해야 합니까?
FDA 승인 날카로운 물건 폐기 용기 또는 적절한 가정용 용기가 없다면 어떻게 해야 합니까?
|
|
| 항상 펜과 날카로운 물건 폐기 용기를 어린이의 손이 닿지 않는 곳에 보관하십시오. | |
|
주사 날짜와 위치를 기록하십시오. HUMIRA를 복용할 때를 기억하는 데 도움이 되도록 미리 달력에 표시하십시오. |
 |
|
| 이 사용 지침은 미국 식품의약국(FDA)의 승인을 받았습니다. | 2023년 11월 개정 |
| AbbVie Inc. North Chicago, IL 60064 U.S.A. 제조 미국 라이선스 번호 1889 20081409 제조사: Cordavis Trading Limited, Dublin, Ireland |
|
사용 지침
| 사용 지침 | |
|
HUMIRA® (휴미라) (adalimumab) 80 mg/0.8 mL, 40 mg/0.4 mL, 20 mg/0.2 mL 및 10 mg/0.1 mL 싱글도즈 사전 충전형 주사기 |
|
주사 전: 의료 서비스 제공자는 귀하가 HUMIRA를 처음 사용하기 전에 사용 방법을 보여주어야 합니다. 도움이 필요하면 의료 서비스 제공자나 1-800-4HUMIRA (1-800-448-6472)에 전화하십시오.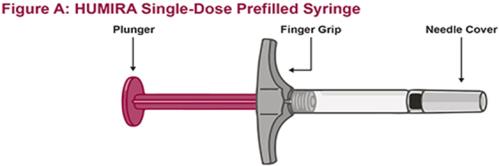 |
|
| HUMIRA 주사 전에 알아야 할 중요 정보 | |
| 다음과 같은 경우에는 사전 충전형 주사기를 사용하지 말고 의료 서비스 제공자나 약사에게 문의하십시오. | |
|
|
| 주사를 맞기 직전까지 바늘 덮개를 씌워 두십시오. HUMIRA는 어떻게 보관해야 합니까? |
|
|
|
|
HUMIRA, 주사 용품 및 다른 모든 의약품은 어린이의 손이 닿지 않는 곳에 보관하십시오. HUMIRA 싱글도즈 사전 충전형 주사기를 사용하기 전에 모든 페이지의 지침을 읽으십시오. |
|
|
HUMIRA를 냉장고에서 꺼냅니다. 주사하기 전에 HUMIRA를 실온에서 15~30분 동안 두십시오.
|
 |
사전 충전형 주사기 라벨에 있는 유효 기간을 확인하십시오. 유효 기간이 지난 경우에는 사전 충전형 주사기를 사용하지 마십시오. 깨끗하고 평평한 표면에 다음을 놓습니다.
손을 씻고 말리십시오. |
 |
주사 부위를 선택합니다.
알코올 솜으로 주사 부위를 원을 그리며 닦습니다.
|
 |
한 손으로 사전 충전형 주사기를 잡습니다. 다른 손으로 바늘 덮개를 똑바로 부드럽게 당겨 빼냅니다.
|
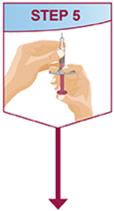 |
바늘이 위로 향하게 하여 사전 충전형 주사기를 잡습니다.
|
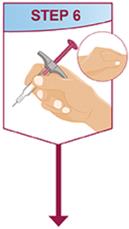 |
엄지와 검지 사이로 한 손으로 사전 충전형 주사기의 몸체를 잡습니다. 연필처럼 손에 사전 충전형 주사기를 잡습니다. 어떤 경우에도 플런저를 뒤로 당기지 마십시오. |
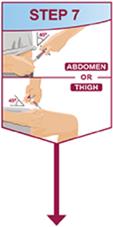 |
다른 손으로 주사 부위의 소독한 피부를 부드럽게 잡아당깁니다. 피부를 단단히 잡습니다. 빠르고 짧게 던지는 듯한 동작으로 바늘을 피부에 약 45도 각도로 삽입합니다.
플런저를 천천히 끝까지 밀어 넣어 모든 액체가 주입되고 사전 충전형 주사기가 비워지도록 합니다. |
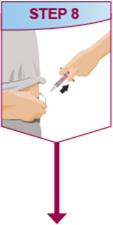 |
주사가 완료되면 사전 충전형 주사기를 같은 각도로 유지하면서 바늘을 피부에서 천천히 빼냅니다. 주사를 완료한 후 주사 부위의 피부에 솜이나 거즈를 놓습니다.
|
 |
사용한 HUMIRA 사전 충전형 주사기는 어떻게 폐기해야 합니까?
|
| 바늘 덮개, 알코올 솜, 솜이나 거즈, 복용량 트레이 및 포장은 일반 쓰레기통에 버려도 됩니다. | |
|
HUMIRA 일회용 사전 충전형 주사기 사용에 대한 질문 의료 서비스 제공자로부터 직접 교육을 받지 못한 경우 어떻게 해야 합니까?
FDA 승인을 받은 뾰족한 물건 폐기 용기나 적절한 가정용 용기가 없는 경우 어떻게 해야 합니까?
|
|
| 항상 사전 충전형 주사기와 뾰족한 물건 폐기 용기를 어린이의 손이 닿지 않는 곳에 보관하십시오. | |
|
주사 날짜와 위치를 기록해 두십시오. HUMIRA 복용 시기를 기억하는 데 도움이 되도록 달력에 미리 표시해 두십시오. |
 |
|
| 이 사용 설명서는 미국 식품의약국(FDA)의 승인을 받았습니다. | 2023년 11월 개정 |
| 제조: AbbVie Inc. North Chicago, IL 60064 U.S.A. 미국 라이선스 번호 1889 20082850 제조 판매원: Cordavis Trading Limited, Dublin, Ireland |
|
주요 표시면
주요 표시면
NDC: 83457-554-02
2개입 일회용 사전 충전형 펜
HUMIRA® PEN
adalimumab
40 mg/0.4 mL
피하 주사용으로만 사용하십시오
29 게이지 바늘
약사 주의: 각 환자에게는 동봉된
복약 안내서를 제공해야 합니다.
전체 상자를 하나의 단위로 조제해야 합니다.
복용량 트레이 씰이 파손되었거나 없는 경우 약국에 반품하십시오.
이 상자에 포함된 내용물:
- 2개의 복용량 트레이(각 트레이에는 29게이지 ½인치 길이의 고정 바늘이 있는 일회용 사전 충전형 펜 1개 포함)
- 2개의 알코올 솜 • 1개의 복약 안내서 • 1개의 포장 첨부 문서 • 1개의 사용 설명서
제조:
cordavis™
HUMIRA.COM
Rx only
abbvie

주요 표시 부분
NDC: 83457-124-02
2개입 1회 투여용 프리필드 펜
HUMIRA® PEN
아달리무맙
80 mg/0.8 mL
피하 주사용
29 게이지 바늘
약사 주의 사항: 각 환자는 동봉된
복약 안내서를 받아야 합니다.
주사기의 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다.
전체 상자를 하나의 단위로 조제해야 합니다.
투여 트레이 씰이 파손되었거나 없는 경우 약국에 반품하십시오.
이 상자의 내용물:
- 2개의 투여 트레이(각 트레이에는 29게이지 ½인치 길이의 고정 바늘이 있는 1회 투여용 프리필드 펜 1개 포함)
- 알코올 솜 2개 • 복약 안내서 1개 • 포장 첨부 문서 1개 • 사용 설명서 1개
제조:
cordavis™
HUMIRA.COM
처방전 의약품
abbvie
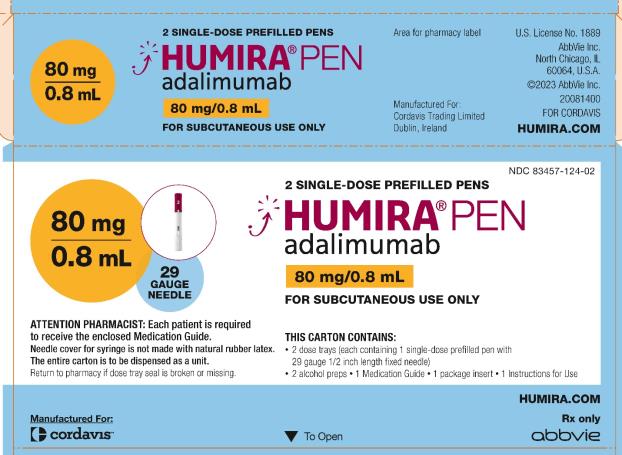
주요 표시 부분
NDC: 83457-817-02
2개입 일회용 사전 충전형 주사기
HUMIRA®
adalimumab
10 mg/0.1 mL
피하 주사용
29 게이지 바늘
약사 주의: 각 환자는 동봉된
복약 안내서를 받아야 합니다.
주사기의 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다.
전체 상자를 하나의 단위로 취급해야 합니다.
복용량 트레이 씰이 파손되었거나 누락된 경우 약국에 반품하십시오.
이 상자에 포함된 내용물:
- 2개의 복용량 트레이(각 트레이에는 29게이지 ½인치 길이의 고정 바늘이 있는 일회용 사전 충전형 펜 1개 포함)
- 2개의 알코올 솜 • 1개의 복약 안내서 • 1개의 포장 삽입물
- 1개의 사용 설명서
제조:
cordavis™
HUMIRA.COM
처방전 필요
abbvie

주요 표시 패널
NDC: 83457-616-02
2개의 단일 용량 사전 충전 주사기
HUMIRA®
adalimumab
20 mg/0.2 mL
피하 주사 전용
29 게이지 바늘
주의 약사: 각 환자는 동봉된
약물 안내서를 받아야 합니다.
주사기용 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다.
전체 상자는 단위로 제공되어야 합니다.
용량 트레이 씰이 깨지거나 누락된 경우 약국으로 반환하십시오.
이 상자에는 다음이 포함되어 있습니다.
- 2개의 용량 트레이(각각 29 게이지 ½ 인치 길이 고정 바늘이 있는 단일 용량 사전 충전 펜 1개 포함)
- 2개의 알코올 소독솜 • 1개의 약물 안내서 • 1개의 포장 삽입물
- 1개의 사용 설명서
제조사:
cordavis™
HUMIRA.COM
처방전에 의해서만 구입 가능
abbvie

주요 표시면
NDC: 83457-243-02
2개입 일회용 사전 충전형 주사기
HUMIRA®
adalimumab
40 mg/0.4 mL
피하 주사용
29 게이지 바늘
약사 주의: 각 환자는 동봉된
복약 안내서를 받아야 합니다.
주사기의 바늘 덮개는 천연 고무 라텍스로 만들어지지 않았습니다
이 상자의 내용물:
- 2개의 투여 트레이(각 트레이에는 29게이지 ½인치 길이의 고정 바늘이 있는 일회용 사전 충전형 펜 1개 포함)
- 알코올 솜 2개 • 복약 안내서 1개 • 사용 설명서 1개
- 사용 지침 1개
제조:
cordavis™
HUMIRA.COM
처방전 필요
abbvie
